
Abstract
The HIV-1 epidemic in Russia is dominated by the former Soviet Union subtype A (AFSU) variant, but other genetic forms are circulating in the country. One is the recently described CRF63_02A1, derived from recombination between a CRF02_AG variant circulating in Central Asia and AFSU, which has spread in the Novosibirsk region, Siberia. Here we phylogenetically analyze pol and env segments from 24 HIV-1 samples from the Novosibirsk region collected in 2013, with characterization of three new near full-length genome CRF63_02A1 sequences, and estimate the time of the most recent common ancestor (tMRCA) and the demographic growth of CRF63_02A1 using a Bayesian method. The analyses revealed that CRF63_02A1 is highly predominant in the Novosibirsk region (81.2% in pol sequences) and is transmitted both among injecting drug users and by heterosexual contact. Similarity searches with database sequences combined with phylogenetic analyses show that CRF63_02A1 is circulating in East Kazakhstan and the Eastern area of Russia bordering China. The analyses of near full-length genome sequences show that its mosaic structure is more complex than reported, with 18 breakpoints. The tMRCA of CRF63_02A1 was estimated around 2006, with exponential growth in 2008–2009 and subsequent stabilization. These results provide new insights into the molecular epidemiology, phylogeny, and phylodynamics of CRF63_02A1.
R
In 1995–1996 HIV-1 infections began to increase dramatically concomitantly with the expansion of a subtype A variant of Central African ancestry 5 that originated in Southern Ukraine 6,7 and spread to all countries of the former Soviet Union (FSU), in most of which it is the predominant HIV-1 genetic form 8,9 ; for this reason it is frequently designated AFSU variant. In addition to AFSU, other HIV-1 genetic forms circulating in FSU countries at lower prevalences include subtype B, predominant in men who have sex with men, 8 –10 CRF03_AB, predominant in the Russian cities of Kaliningrad 11 and Cherepovets, 12 subtype F, circulating as a minor variant in St. Petersburg, Russia, 13 and a CRF02_AG variant (CRF02_AGFSU), 14,15 which was first detected among injecting drug users (IDUs) in Tashkent, Uzbekistan, in 1999–2000, 14 and has subsequently been reported in Kazakhstan, 15 Kyrgyzstan, 16 and in the Novosibirsk region, Siberia, Russia. 17 This variant has generated, through secondary recombination with AFSU, a new circulating recombinant form, CRF63_02A1, recently identified in Novosibirsk. 17,18 Here we examine the prevalence of CRF63_02A1 in the Novosibirsk region in samples collected in 2013, we obtain three new near full-length genome sequences of CRF63_02A1, reanalyzing its mosaic structure, and we estimate its epidemic history.
For this study, 26 serum samples from HIV-1-infected individuals were collected in May and June 2013 at the Center for Prevention and Control of AIDS and Infectious Diseases, Koltsovo, Novosibirsk region, which is located 5 km from the city of Novosibirsk and attends all HIV-infected people of the Novosibirsk region. This study was approved by the Ethics Committee of the Center for Prevention and Control of AIDS and Infectious Diseases at Koltsovo, Novosibirsk region, Russia.
RNA was extracted from 1 ml plasma using Nuclisens EasyMAG kit (bioMérieux, Marcy l'Etoile, France) following the manufacturer's instructions. The HIV-1 protease-reverse transcriptase (PR-RT) segment of pol and the C2-V3-C3 segment of env were amplified by reverse transcriptase polymerase chain reaction (RT-PCR) followed by nested PCR. Primers used for PR-RT amplification were RP1-S and RP-1-A in RT-PCR and PR-O-S2b and RT-O-A in nested PCR (Table 1) and those used for amplification of the V3 region were described previously. 19 Near full-length genome (∼9 kb) amplification in overlapping segments by RT-PCR and nested PCR and sequencing was done using a protocol similar to that described by us, 20,21 although some amplified fragments and their corresponding primers, all of which are listed in Table 1, were different. For amplification of the 3′ semigenome, primers SG3-up and SG3-lo were used for RT-PCR. This would allow for amplification of the full-length envelope sequence in nested PCR using primers Env-S and Env-A, which could be used for subsequent construction of functional envelope clones. 22
S, sense; A, antisense.
Both fragments flanking env, from vif through vpu, and nef plus a segment of the 3′ LTR, respectively, were amplified by nested PCR from the RT-PCR product amplified with SG3-up and SG3-lo, using primers SG3-N-S and SSD2b, and NEF-S4 and 3′ nef-3c, respectively. Gag and a fragment of the 5′ LTR were amplified with SC-A-O-S-R and SC-A-O-A in RT-PCR and SC-A-N-S-R and SC-A-N-A in nested PCR; the 3′ segment of pol was amplified with RTDS-O-S and SC-B-N-A in RT-PCR and RTDS-N-S and B2-N-A in nested PCR. Sequence electropherograms were assembled with Seqman (DNASTAR, Madison, WI).
Sequences were aligned with MAFFT v.7. 23 Phylogenetic trees were constructed via maximum likelihood with RAxML v.7.2.7, 24 applying the general time reversible (GTR) substitution model with CAT approximation for among-site rate heterogeneity (GTR+CAT), with assessment of node support by bootstrapping. Recombination was analyzed by bootscanning with SimPlot v3.5, 25 using a 200 nucleotide (nt) window with tree construction by the neighbor-joining method applying Kimura's two-parameter substitution model (the shorter than usual window size employed in the analysis was intended for a more precise definition of recombinant structures, with detection of fragments delimited by relatively close breakpoints).
Short putatively recombinant segments (≤200 nt) identified with SimPlot were phylogenetically analyzed further via maximum likelihood (ML) using RAxML, applying the GTR+CAT model, and with PhyML v3.0, 26 applying the GTR with gamma-distributed rate heterogeneity among sites and a proportion of invariable sites (GTR+G+I) substitution model, with assessment of node support by the approximate likelihood ratio test (aLRT) using a Shimodaira–Hasegawa (SH)-like procedure. In this analysis, trees were constructed with reference sequences of CRF02_AGFSU and AFSU and were rooted with an inferred group M ancestral sequence downloaded from the HIV Sequence Database. 27 Segments were assigned to either variant based on clustering with a bootstrap value ≥70% with RAxML or an aLRT-SH-like support ≥0.8 with PhyML.
To identify CRF63_02A1 viruses from other geographic areas, we downloaded all HIV-1 sequences from FSU countries deposited in the HIV Sequence Database.
27
The similarity of these sequences to all available CRF63_02A1 and CRF02_AG near full-length genome sequences was examined with local BLAST searches using BioEdit v.7.1.3.0 (Tom Hall,
Antiretroviral (ARV) drug resistance in PR-RT sequences was analyzed with the online Stanford University HIV Drug Resistance Database's HIVdb Program. 28
To estimate the time of the most recent common ancestor (tMRCA) and to analyze the demographic growth of CRF63_02A1, we used a Bayesian Markov chain Monte Carlo (MCMC) coalescent method as implemented in BEAST v1.7.4. 29 For these analyses, we used 105 CRF63_02A1 PR-RT sequences lacking drug resistance mutations, including 16 obtained by us and 89 downloaded from the HIV Sequence Database, collected between 2009 and 2013, as well as three CRF02_AGFSU sequences used as outgroups. Since the evolutionary rate could not be inferred directly from CRF63_02A1 sequences, due to the narrow time span of sample collection, it was estimated using 100 A1 subsubtype PR-RT sequences (considering that most of the analyzed sequence is of A1 subsubtype) sampled along a time span of 23 years and lacking drug resistance-associated mutations, retrieved from the HIV Sequence Database. In the subsequent analysis, the estimated posterior distribution of the substitution rate was incorporated as a prior distribution in the analysis of CRF63_02A1 sequences. The substitution model used for these analyses was HKY with gamma-distributed rate heterogeneity among sites and two partitions in the codon positions (first+second, third); other priors were an uncorrelated lognormal relaxed clock model and a Bayesian skyline plot demographic model. Each MCMC chain was run for 50 million generations, sampling every 1,000 generations, with the first 10% discarded as burn-in. MCMC convergence and effective samples sizes were checked using the program Tracer v.1.5.
At least one genome fragment could be sequenced in 24 HIV-1 samples from Novosibirsk: 16 in both PR-RT and the V3 region, six only in PR-RT, and two only in V3. Demographic and clinical data of these samples are shown in Table 2. Phylogenetic trees of PR-RT and V3 segments are shown in Fig. 1a and b, respectively. Phylogenetic classification according to these analyses was incorporated in Table 2. In PR-RT, where CRF63_02A1 viruses grouped in a monophyletic clade closely related to CRF02_AGFSU viruses, 18 (81.8%) samples were CRF63_02A1 and four (18.2%) were AFSU (Fig. 1a). In the V3 region, where CRF63_02A1 viruses did not group in a separate clade, but branched in a clade together with CRF02_AGFSU viruses, 17 (94.4%) samples branched in the CRF02_AGFSU/CRF63_02A1 clade and one (5.6%) was AFSU (Fig. 1b). In the 16 samples in which both segments could be sequenced, 14 (87.5%) were CRF63_02A1 in both segments, one, RU_8508, was AFSU in both segments, and one, RU_8506, had incongruent topologies, being AFSU in PR-RT and CRF63_02A1 in V3, suggesting the presence of recombination between both variants.
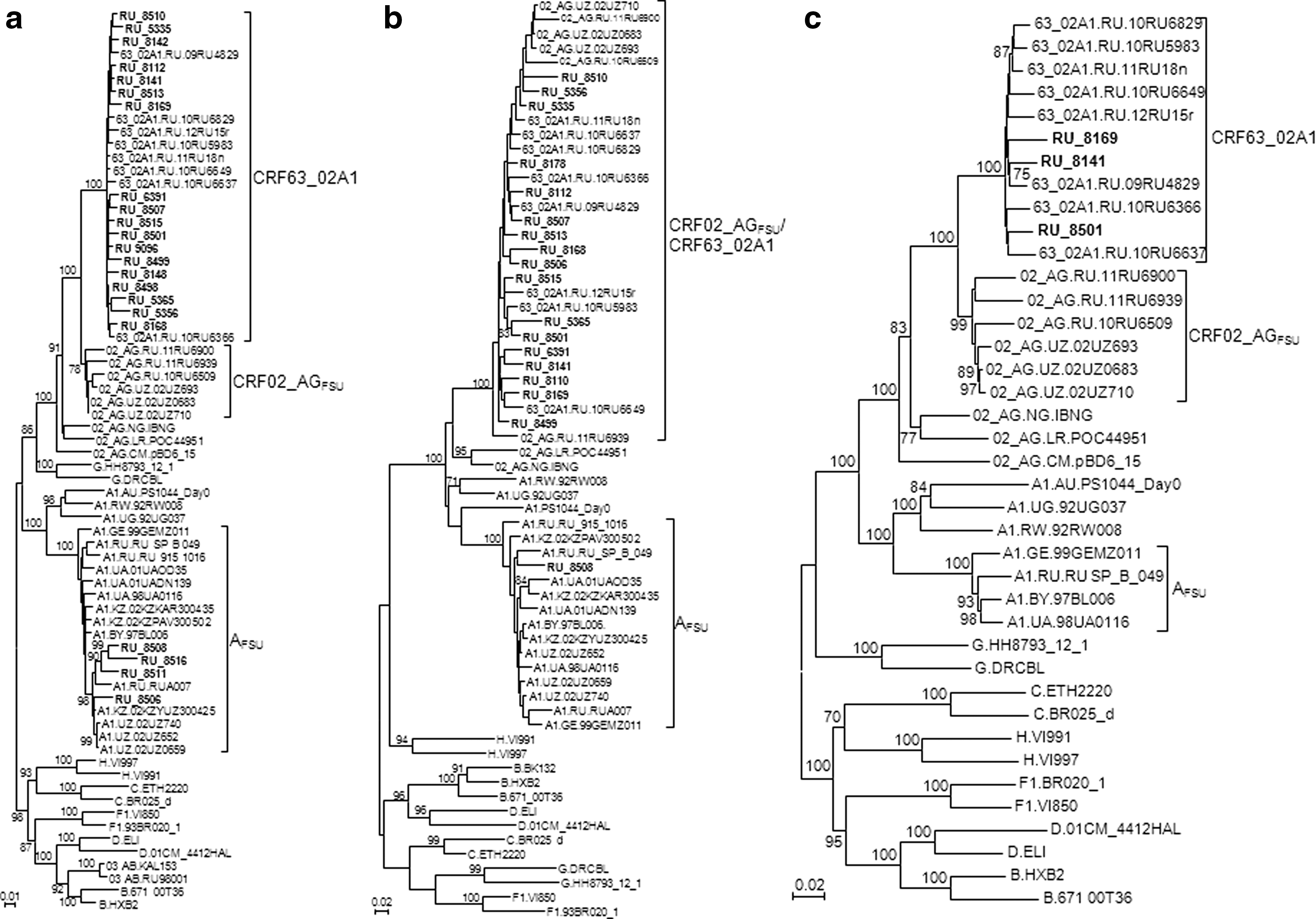
Phylogenetic trees of HIV-1 sequences from samples from Novosibirsk.
IDU, injecting drug user; Het, heterosexual; Sex, sexual.
n.a., datum not available; ARV, antiretroviral; CRF, circulating recombinant form; PR-RT, protease reverse transcriptase.
Of the 20 samples in which at least one segment was of CRF63_02A1 and data on transmission route were available, 13 (65%) corresponded to IDUs and seven (35%) to heterosexual transmissions. The identification of CRF63_02A1 both among IDUs and heterosexually infected individuals is similar to what has been reported with regard to AFSU in Russia. 8,9 Epidemiological data indicate that heterosexual transmission of HIV-1 in FSU countries is closely linked to the epidemic among IDUs, 1,30 which is consistent with our studies in St. Petersburg showing that infections among IDUs and heterosexually infected individuals were not associated with different AFSU clusters. 10 With regard to CRF63_02A1, analyses of a greater number of samples with epidemiological data would be required to examine the existence of transmission networks and their possible associations with different transmission routes.
Antiretroviral (ARV) drug resistance-associated mutations were found in three samples, RU_8148, RU_8499, and RU_8516. The first had low degree resistance to nucleoside RT inhibitors (M41L), the second had high or intermediate resistance to nonnucleoside RT inhibitors (K103N), and the third had resistance to both drug classes (D67N, K70R, M184V; K101E, G190S). RU_8148 and RU_8516 were on ARV drug treatment and no information on drug treatment was available for RU_8499.
Near full-length genome sequences were obtained in three samples. The phylogenetic tree (Fig. 1c) shows that the newly derived sequences branch within the CRF63_02A1 clade, which is closely related to the CRF02_AGFSU clade. To analyze the mosaic structure of CRF63_02A1, a consensus sequence was obtained using all 11 available CRF63_02A1 near full-length genome sequences, including the three newly derived ones and the eight obtained previously, 17,18 which was used for bootscan analysis. Since there is prior knowledge on the parental strains of CRF63_02A117 we used as references only consensus sequences of AFSU and CRF02_AGFSU, with consensuses of subtypes C and H being used as outgroups. The plot derived from this analysis (Fig. 2a) suggests a complex recombinant structure with most of the genome derived from CRF02_AGFSU. Short segments (<200 nt) in which clustering with either AFSU or CRF02_AGFSU consensuses was supported by ≥50% bootstrap values were further analyzed via ML with RAxML and PhyML (Fig. 2b). These analyses made it possible to assign eight short segments to either variant clustering with the respective reference sequences with ≥70% bootstrap and/or ≥0.8 aLRT-SH-like support values.
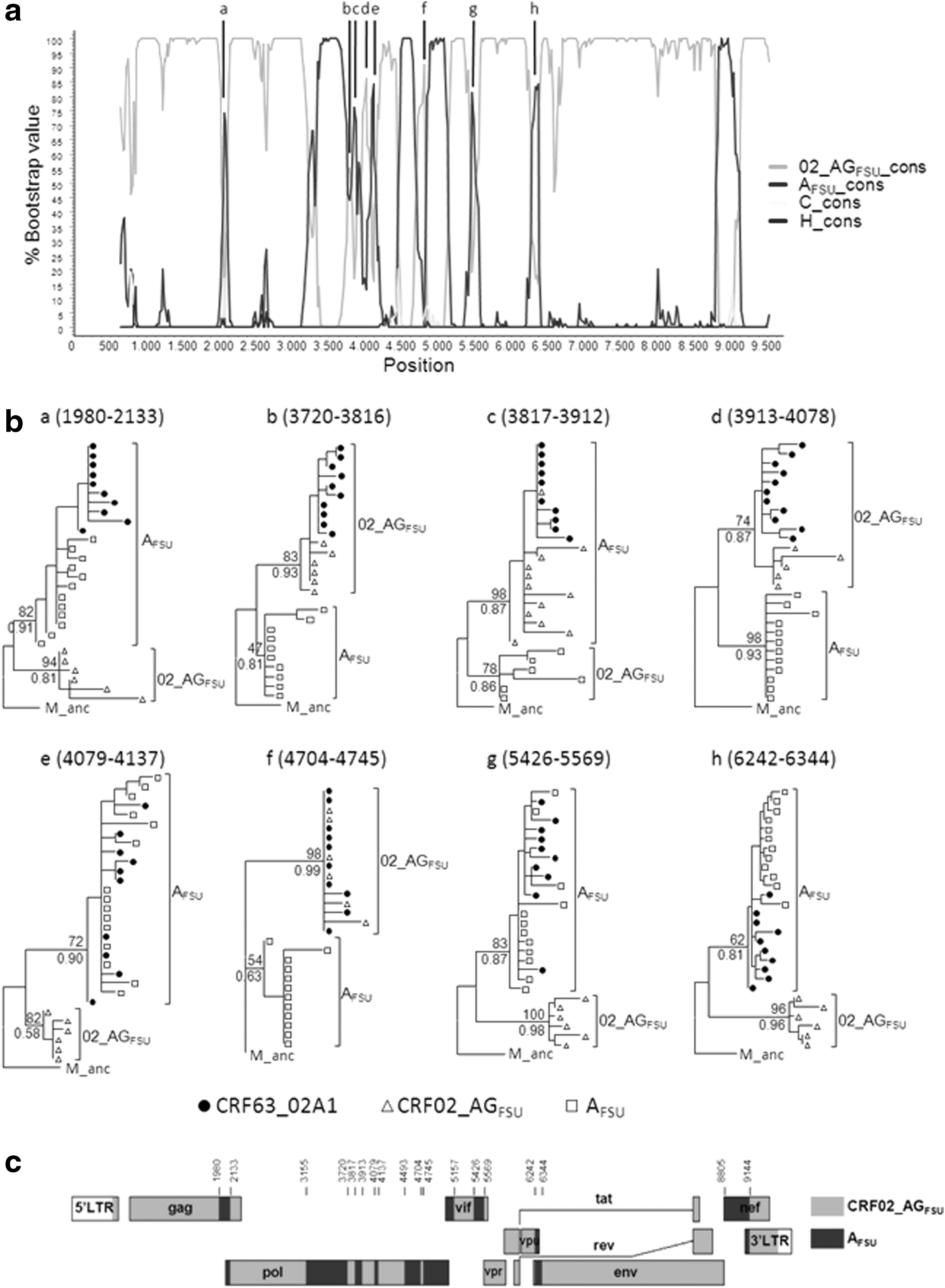
Analysis of the mosaic structure of CRF63_02A1.
The mosaic structure of CRF63_02A1 derived from the bootscan analysis with SimPlot combined with the ML trees of partial segments (Fig. 2c) shows the existence of 18 breakpoints delimiting 10 CRF02_AGFSU-derived segments and nine AFSU-derived segments. The inferred structure is much more complex than what was previously described, 17 in which only five AFSU-derived segments were identified. The difference may derive from the different methods of analysis and from the use as references in our study of the parental strains of CRF63_02A1 (AFSU and CRF02_AGFSU), which may allow for a more precise definition of the recombinant structure than when using more distantly related references.
To identify CRF63_02A1 sequences deposited in the Los Alamos HIV Sequence Database, CRF63_02A1 PR-RT sequences were used for BLAST searches with subsequent phylogenetic analyses, as described in the Materials and Methods section. This analysis revealed that in addition to being found in Novosibirsk, where they represented 59.9% of database sequences from this region collected in 2009–2013 (results not shown), CRF63_02A1 viruses were found in the Eastern area of Russia, near the border with China, in the cities of Khabarovsk (13 of 88 sequences, 14.8%) and Blagoveshchensk (2 of 40 sequences, 5%), and in the Ust-Kamenogorsk region in East Kazakhstan (3 of 14 sequences, 21.4%) (Fig. 3). This is in agreement with recent studies reporting the identification of CRF63_02A1 in the Russian Far East 31 and Kazakhstan. 32 However, we could not find CRF63_02A1 among database viruses from Kyrgysztan, as suggested by others. 17
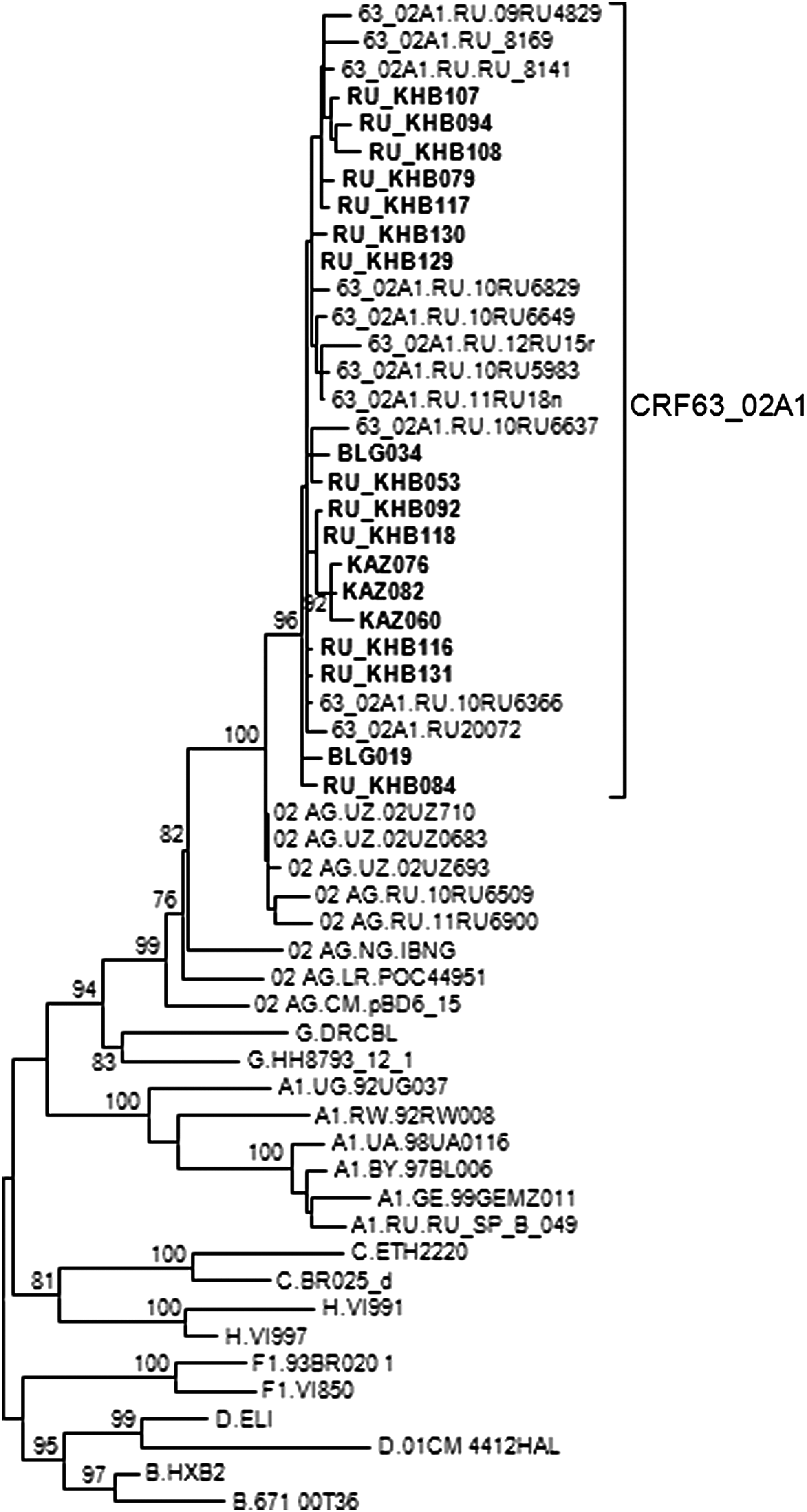
Phylogenetic tree of PR-RT sequences showing viruses from East Kazakhstan and the Eastern Russian cities of Khabarovsk and Blagoveshchensk branching within the CRF63_02A1 clade. The names of the mentioned viruses, which include the KAZ, KHA, and BLG abbreviations of the sampling locations, are in bold type. Only bootstrap values ≥70% are shown.
Through Bayesian MCMC analyses using PR-RT sequences obtained by us and deposited in databases, the tMRCA of CRF63_02A1 was estimated in 2006.8 [95% highest posterior density (HPD) interval 2005.8–2007.7]. Demographic growth estimated through the Bayesian Skyline Plot shows an exponential increase in the effective number of infections during a period comprising most of 2008 and the first half of 2009, with subsequent stabilization (Fig. 4). These results are consistent with the earliest known date of HIV diagnosis of a CRF63_02A1 infection in 2007 (Table 1) and of the collection of the first sample harboring a CRF63_02A1 virus in 2009. 27 They are also in accord with the largest annual increase in new HIV-1 diagnoses recorded in the Novosibirsk region, which occurred in 2008 (Fig. 5).
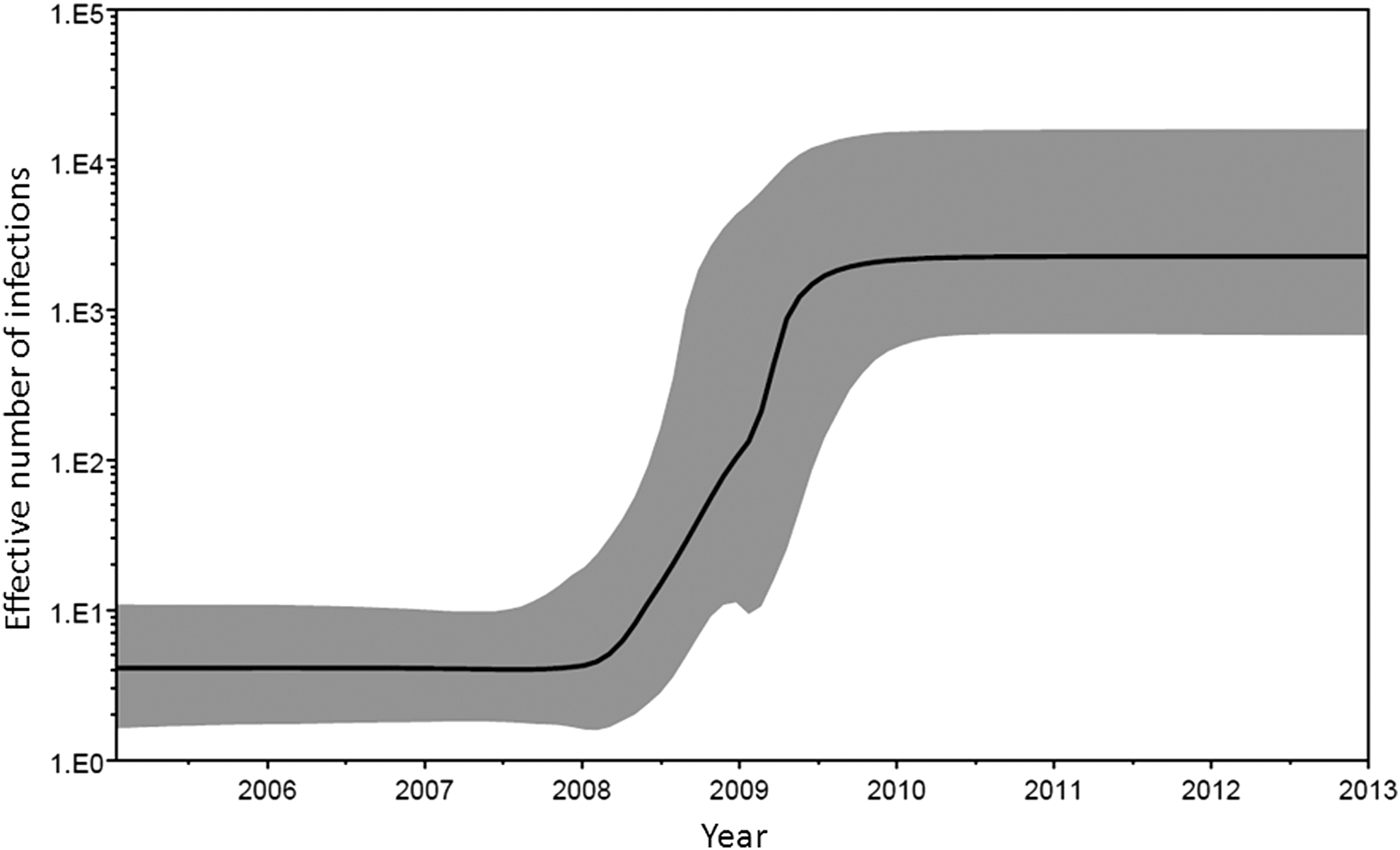
Bayesian skyline plot of the population growth of CRF63_02A1. The plot was obtained using PR-RT sequences. The black line represents the median estimate of the effective number of infections through time, plotted on a logarithmic scale, and the shaded area represents the 95% HPD confidence interval for this estimate.
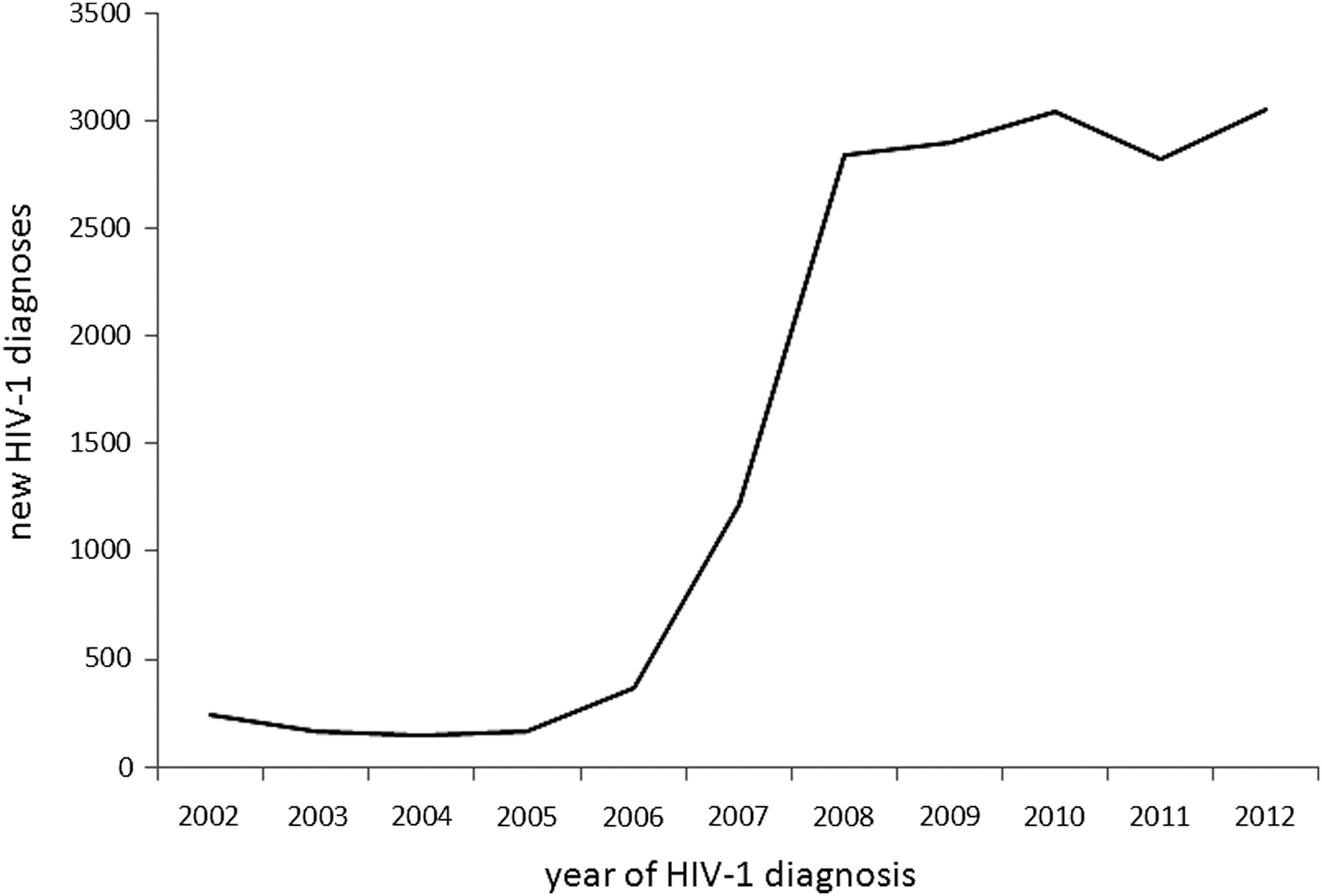
Time trend in new HIV-1 diagnoses in the Novosibirsk region (2002–2012). Data are from the Center for Prevention and Control of AIDS and Infectious Diseases, Koltsovo, Novosibirsk region, Russia (
In summary, this study shows that CRF63_02A1 is highly predominant in recently collected samples in Novosibirsk, where it is transmitted both among IDUs and via heterosexual contact and that its mosaic structure is much more complex than previously reported. CRF63_02A1 originated recently and expanded rapidly, with most of its expansion taking place along an approximate period of only one and a half years (Fig. 4). The rapid expansion of CRF63_02A1 is reminiscent of that reported for other HIV-1 genetic forms in established epidemics, 33,34 supporting the need for continued molecular epidemiological surveillance of HIV-1. Its propagation to distant areas of Russia and to Kazakhstan indicates that CRF63_02A1 should be taken into consideration in the design of vaccines adapted to HIV-1 strains circulating in Russia and other FSU countries, whose HIV-1 epidemics are becoming increasingly complex by the introduction of new genetic forms, phylogenetic diversification within the established strains, 10,35 –37 and recombination between cocirculating variants, with the generation of new circulating recombinant forms (CRFs). 17
Sequences were deposited in GenBank under accession numbers KJ197185–KJ197224.
Footnotes
Acknowledgments
We thank the personnel at the Genomic Unit of Instituto de Salud Carlos III, Majadahonda, Madrid, Spain, for technical assistance in sequencing, and Bonnie Mathieson, from the Office of AIDS Research, National Institutes of Health, Bethesda, Maryland for her support of this study. This work was funded by Office of AIDS Research, National Institutes of Health, through the training program “Molecular Epidemiology of HIV-1 in Eastern Europe and Its Significance for Vaccine Development.”
Author Disclosure Statement
No competing financial interests exist.