
Abstract
The presence of infection by human immunodeficiency virus type 2 (HIV-2) in Cuba has been previously documented. However, genetic information on the strains that circulate in the Cuban people is still unknown. The present work constitutes the first study concerning the phylogenetic relationship of HIV-2 Cuban isolates conducted on 13 Cuban patients who were diagnosed with HIV-2. The env sequences were analyzed for the construction of a phylogenetic tree with reference sequences of HIV-2. Phylogenetic analysis of the env gene showed that all the Cuban sequences clustered in group A of HIV-2. The analysis indicated several independent introductions of HIV-2 into Cuba. The results of the study will reinforce the program on the epidemiological surveillance of the infection in Cuba and make possible further molecular evolutionary studies.
T
Phylogenetic analysis of HIV-2 sequences has resulted in seven groups being described (A–G), with a predominance of A and B. HIV-2 group A is the predominant group circulating in West Africa (Senegal and Guinea Bissau) and HIV-2 group B predominates in the Ivory Coast. The other groups have been reported in only a few individuals. Groups C, D, E, and F were found in rural areas of Sierra Leone and Liberia and group G in the Ivory Coast. 7 In HIV-2-infected individuals the presence of a circulating recombinant form (CRF), 01_AB, has been detected. 8,9
In Cuba, from 1986 to December 2013, a total of 20 individuals were serologically confirmed as positive for antibodies to HIV-2 in the AIDS Research Laboratory (National Reference Laboratory for Human Retroviruses). 10,11 However, there are no reports available about HIV -2 groups circulating in the Cuban population. The aim of this study was to characterize the genetic diversity of HIV-2 in Cuban patients.
In 2012 a total of 13 HIV-2-infected individuals were investigated and a 10-ml EDTA blood sample was taken; this was done after obtaining a written informed consent and the recording of epidemiological data, including age, sex, sexual behavior, and place of infection (Table 1). Plasma samples were screened using ELISA DAVIH-HIV-2 (DAVIH Laboratories, Cuba) according to the manufacturer's specifications. 11 The positive samples were confirmed by using a Western blot (DAVIH-blot HIV-2, DAVIH Laboratories, Cuba) assay following the manufacturer's instructions. 11
M, male; F, female; MSM, men who have sex with men; HT, heterosexual.
The proviral DNA was extracted from whole blood using the High Pure Viral Nucleic Acid kit (Roche Diagnostics, IN) according to the manufacturer's instructions. The env gene of HIV-2 was amplified by a nested polymerase chain reaction (PCR) “in house” with the external primers 57444 (5′- AATAGRGATACTTGGGGRAC-3′) (SMM239 genome 6745 to 6764) and 55901 (5′-TTCTGTTYCTTCKYTGGYTGGC-3′) (SMM239 genome 8853 to 8832) and inner primers 57443 (5′-AGTAACAGAACARGCARTAGA-3′) (SMM239 genome 6846 to 6866) and 55903 (5′-GGARGAGAAMACWGGCCKATA-3′) (SMM239 genome 8784 to 8764). The PCR reaction was performed with the use of the Fast Start High Fidelity PCR System (Roche Diagnostics, GmbH, Mannheim, Germany) with the following cycling conditions: first round (2 min at 94°C, 39 cycles of 15 s at 95°C, 30 s at 50°C, and 3 min at 65°C, with an extension of 7 min at 72°C) and second round (1 min at 94°C, 39 cycles of 15 s at 94°C, 30 s at 50°C, and 2 min at 72°C, with an extension of 7 min at 72°C). The PCR product was 1,897 bp. Sequencing in both directions was performed using the Genome Lab Dye Terminator Cycle Sequence with Quick Start kit following the manufacturer's directions (Beckman Coulter, Inc., Fullerton, CA). Sequencing products were read on a CEQ 8800 Genetic Analyzer (Beckman Coulter, Inc., Fullerton, CA).
Cuban sequences were aligned with the env gene reference sequences group of HIV-2 downloaded from the Los Alamos database (
Of patients, 69.2% acquired the infection through heterosexual contact (Table 1). The phylogenetic analysis of the env gene allowed grouping the 13 patient's nucleotide sequences in HIV-2 group A, with a bootstrap value of 74%. Of sequences, 61.5% were grouped by their homology with sequences coming from Guinea Bissau and Senegal (Cluster I), while the 38.5% of Cuban sequences were clustered with sequences from Guinea Bissau (Cluster II) (Fig. 1).
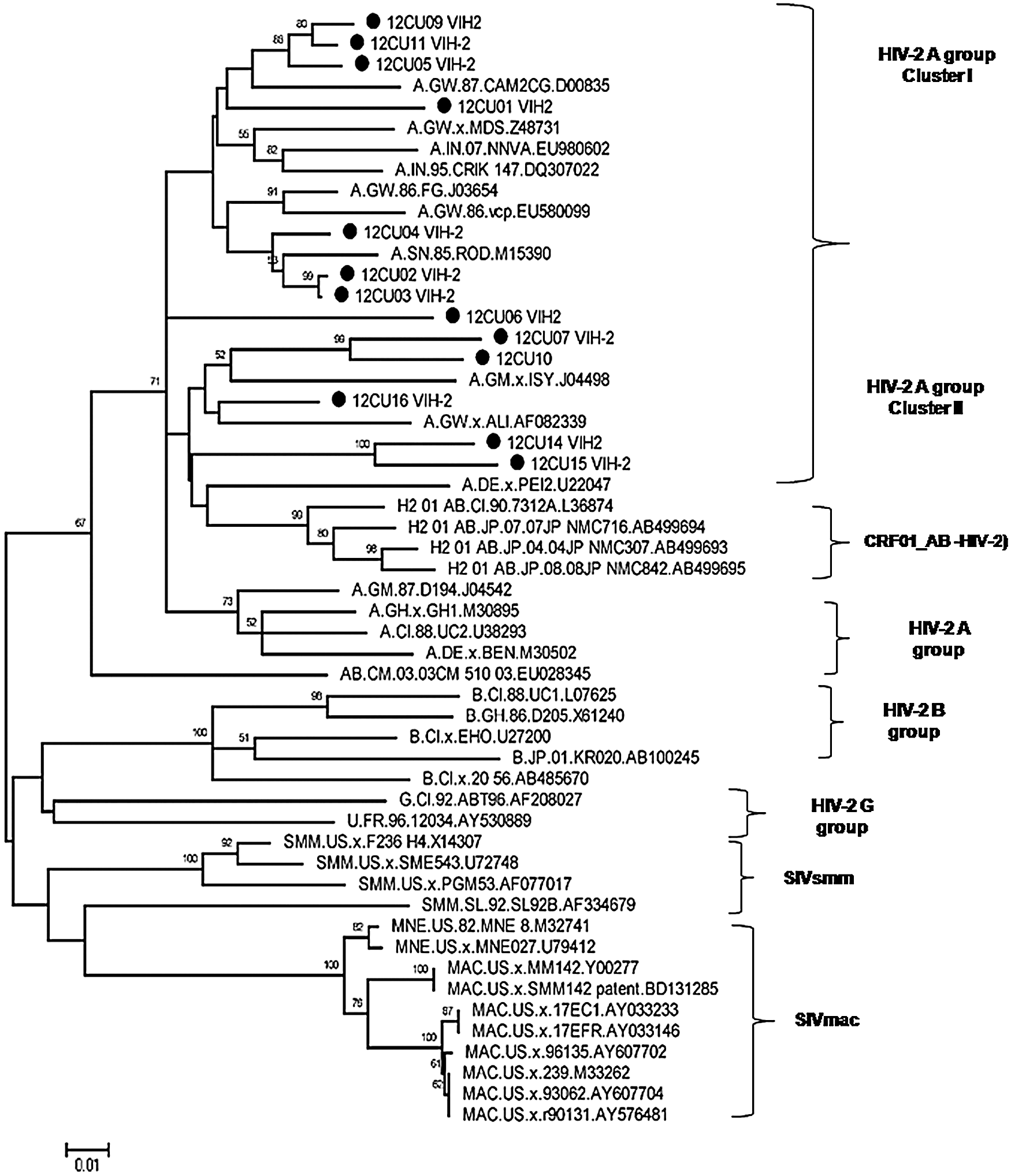
Maximum likelihood (ML) tree of env gene HIV-2 sequences. ML analysis was performed under the HKY+G nucleotide substitution model. All 13 Cuban sequences are indicated by a solid circle (•). Numbers on branches indicate the degree of support for each node.
An essential component of Cuba's National HIV/AIDS Program is the laboratory diagnosis, which makes it possible to differentiate between HIV-1 and HIV-2. 10 Although the presence of HIV-2 in Cuba has already been documented, 10,11 this report is one of the first studies involving circulating HIV-2 genetic variants in the Cuban population.
HIV-2 group A is categorized as an epidemic group and although it is mainly restricted to West Africa, 15 other countries have already reported it, 3 –6 so it is not surprising that all Cuban sequences were found inside this group. Cuban HIV-2 sequences are intermingled with HIV-2 sequences from samples collected in Africa, Japan, Germany, and elsewhere rather than forming a monophyletic clade.
It thus appears that there have been several independent introductions of HIV-2 into Cuba with limited spread of the virus after arrival. There are some subclusters, such as 12CU11–12CU05–12CU09, that likely represent some spread of HIV-2 after its arrival in Cuba. According to epidemiological data obtained concerning the place of infection, most of these patients were infected in countries located in West Africa such as Angola and Cape Verde, countries with a high prevalence of HIV-2. 1,2 It has been reported that group A is the predominant genetic variant in the population infected with HIV-2, mainly in Guinea Bissau, a country with the highest prevalence (8–10%). 16 Also detected was an increase of this viral group in Europe due to the presence of immigrants from West African countries. 3 –5,17
The 12CU015 sample, clustered in Cluster II, corresponding to a patient who acquired the infection in Guinea Bissau, is related to individuals residing in the central (Villa Clara) and eastern regions (Granma and Santiago de Cuba) of Cuba, making it possible to infer the formation of a monophyletic cluster based on place of residence. Similar behavior has been described in a phylogenetic analysis of a group of HIV-2 sequences from individuals from different geographic regions in Italy, which were grouped into different clusters according to country of origin and/or infection. 6
In Cluster I, the sequences from patients infected in Cuba formed a subcluster among them. Patients grouped in this subcluster acquired infection by heterosexual contact and reside in Havana province, suggesting local transmission of this viral group in the HIV-2-positive population. A similar result was reported by Jeong-Gu Nam et al. in 2006, who investigated the molecular subtype of HIV-2 in a group of South Korean-infected individuals. 18
According to the date of diagnosis, epidemiological information, and grouping with sequences from West African countries, Japan, India, and Germany, we can infer that the HIV-2 group A circulating in the HIV-positive Cuban population indicates several independent introductions of HIV-2 into Cuba.
In conclusion, this study shows the presence of HIV-2 group A in Cuba. The results contribute to the program involving the surveillance of HIV-2 and make it possible to conduct studies on its origin, evolutionary trends, and phylogeographic and phylodynamic characteristics.
Sequence Data
GenBank accession numbers for the sequences reported here are from KJ677041 to KJ677053.
Footnotes
Author Disclosure Statement
No competing financial interests exist.