
Abstract

E
To weigh or balance all the data and knowledge in the LANL HIV sequence database, we need appropriate selection criteria. Based on the description of the LANL HIV sequence database, the selection of representative sequences that go into the compendium is a complex process. The current criteria take into account the quality of the sequences, representative genetic diversity, geographic distributions, as well as layout restrictions, with a preference for more prevalent and recently sampled sequences. 1,2 Nevertheless, there still are limitations, regardless of the rationality of the standard itself as well as in practical applications.
We checked the HIV Sequence Compendium for the past 10 years and found that every year the Compendium selects more recently sampled sequences to reflect the current HIV situation. However, we should be more cautious when the “updates” are just transient and unstable cases, for example, a foreign transmission event or a new recombinant form, which have neither a substantial number nor a prevalence trend. In another situation, the “updates” are just minority and regional cases, for example, the representative sequence of subtype B from China has changed from DQ990880 (2008–2011) to HQ215556 (2012) and then to JX140658 (2013). The latter two sequences were sampled more recently and in theory may reflect the recent prevalence of the subtype B pandemic in China.
However, according to the article's description 3 and also our analysis (Fig. 1), HQ215556 and JX140658 are more likely European and American subtype B rather than B′, which is a unique regional variant of subtype B that has caused explosive epidemics in China and Southeast Asia. 4 –7 Therefore, to accurately represent the prevalence of subtype B (B′) in China, it may be better to choose a B′ sequence.
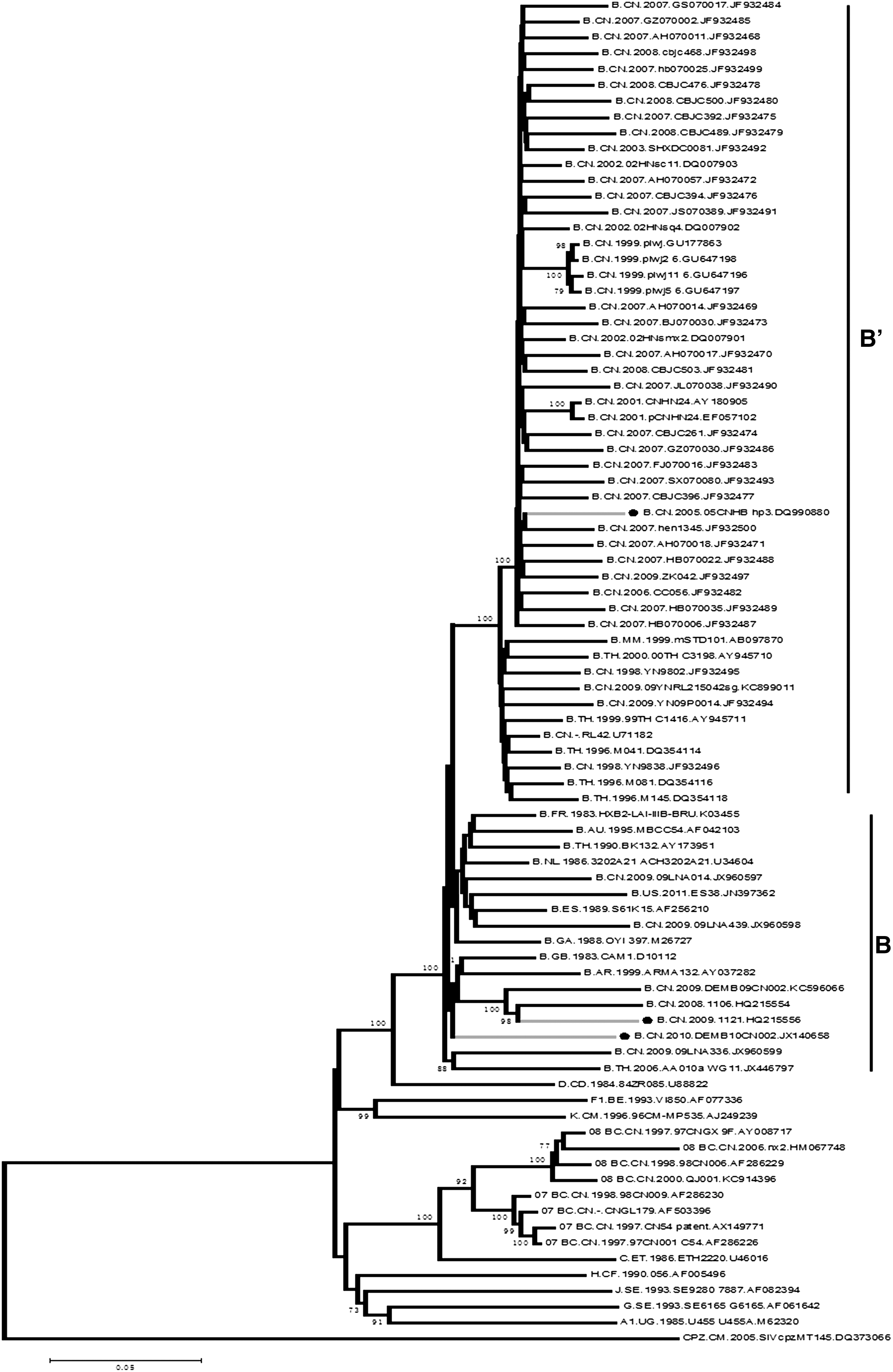
Neighbor-joining tree of full length HIV-1 subtype B sequences from China. Bootstrap values greater than 70 are shown at their corresponding nodes. Filled circles represent the three subtype B representative sequences from China: DQ990880 (2008–2011), HQ215556 (2012), and JX140658 (2013).
In addition, we also noticed that for the most frequent circulating subtypes and circulating recombinant forms (CRFs), only one representative sequence is selected for each country. However, when a subtype or CRF in a region has a complicated genetic background and complicated transmission routes, for example, with some clearly separated prevalence clusters that correlate with different transmission groups, the rigorous selection of one sequence from one country can be relaxed to include more sequences or to include the use of consensus sequences, which may reflect the present situation more accurately.
The current considerations used in selecting compendium sequences also narrow down the selection criteria and the practical applications involved. By means of ongoing discoveries in HIV research, such as epidemiological, biological, and clinical findings, the selection can be optimized so as to compile more rational compendium sequences with the final aim of maximizing its utility in the field of HIV research. The compendium can integrate more additional information or be separated into multilayered sections according to different applications.
As mentioned above, in addition to capturing the genetic variability and geographic distribution of the sequence, transmission networks, the corresponding susceptible populations, and HLA features can also be included to provide increased insight into its evolutionary mechanism and as a means to provide effective surveillance of the prevalence trend. In the case of drug treatment and design, the compendium can also integrate sensitive and resistance polymorphism data, which may indicate the escape mechanism and also provide a quick guide to local drug usage or design. In the case of vaccine design, one of the key considerations is to choose a rational global diversity virus panel to reagent design and broadly neutralizing antibody assessment. 8,9 As little information was available, there is no consistency on how to choose the panel to represent the current status and to assess the results accurately. Taking into consideration sequence variation, sampling time, geographic distribution, clonal sequence sources and transmission routes, glycosylation patterns, cytotoxic T-lymphocytes (CTL) and neutralizing antibody (NAb) epitopes, coreceptor usage, and so on, an optimized representative sequence can be an appropriate candidate for us to use to build such a panel.
Certainly the limitations mentioned above may be due to layout restrictions and its current purpose as a brief printed summary of the HIV sequence database. And in fact the HIV databases provide many useful tools and resources, such as web alignment and various prebuilt alignments for specific purpose. These tools and data are more useful when we deal with the majority of research. Finally we suggest that the compendium be used cautiously; in addition, it should be optimized and extended to encompass a more significant role beyond its current purpose.
Footnotes
Acknowledgments
All the authors contributed to the concept, design, and writing of this article. This study was supported by the Key National Science and Technology Program in the 12th Five-Year Period (grant 2012ZX10001-002) and the Key Laboratory on Emerging Infectious Diseases and Biosafety in Wuhan.
Author Disclosure Statement
No competing financial interests exist.