
Abstract
Studying the genetic diversity and natural polymorphisms of HIV-1 would benefit our understanding of HIV drug resistance (HIVDR) development and predict treatment outcomes. In this study, we have characterized the HIV-1 genetic diversity and natural polymorphisms at the 5′ region of the pol gene encompassing the protease (PR) and reverse transcriptase (RT) from 271 plasma specimens collected in 2008 from HIV-1-infected patients who were eligible for initiating antiretroviral therapy in Abuja (Nigeria). The analysis indicated that the predominant subtype was subtype G (31.0%), followed by CRF02-AG (19.2 %), CRF43-02G (18.5%), and A/CRF36-cpx (11.4%); the remaining (19.9%) were other subtypes and circulating (CRF) and unique (URF) recombinant forms. Recombinant viruses (68.6%) were the major viral strains in the region. Eighty-four subtype G sequences were further mainly classified into two major and two minor clusters; sequences in the two major clusters were closely related to the HIV-1 strains in two of the three major subtype G clusters detected worldwide. Those in the two minor clusters appear to be new subtype G strains circulating only in Abuja. The pretreatment DR prevalence was <3%; however, numerous natural polymorphisms were present. Eleven polymorphic mutations (G16E, K20I, L23P, E35D, M36I, N37D/S/T, R57K, L63P, and V82I) were detected in the PR that were subtype or CRF specific while only three mutations (D123N, I135T, and I135V) were identified in the RT. Overall, this study indicates an evolving HIV-1 epidemic in Abuja with recombinant viruses becoming the dominant strains and the emergence of new subtype G strains; pretreatment HIVDR was low and the occurrence of natural polymorphism in the PR region was subtype or CRF dependent.
Introduction
O
In Nigeria, studies have shown a diversified HIV-1 epidemic with the viral subtype G, CRF06-cpx, CRF02-AG, sub-subtype A3, and other recombinants cocirculating. 16,18,34,38 –40 In a study published in 2000, subtype A was predominant (about 70%) in the southwest-Lagos state and subtype G was predominant in the northwest-Kano state (about 58%), while both subtypes A (49%) and G (47%) were observed to be equally distributed in the northeast (Maiduguri). 18 In 2006, a study in Oyo state (southeast) showed the predominance of CRF02-AG (57%), subtype G (26%), and CRF06-cpx (11%), 16 and similar results with 39–45% for CRF02-AG and 38% for subtype G were reported in 200941 and 2012. 39
Characterization of the polymorphisms within the protease (PR) and reverse transcriptase (RT) genes have been conducted mostly for subtype B viruses; few studies have been conducted for non-B subtype viruses, and their impact on highly active antiretroviral therapy (HAART) is undetermined. 9,29,42 –46 Indeed, it has been shown that differences in codon sequences at positions associated with drug resistance mutations (DRMs) might predispose viral isolates of different subtypes to encode different amino acid substitutions that can affect the rate of emergence of resistance, cross-resistance to same-class drugs, and potentially drug susceptibility and clinical outcomes. 8,47 Data from virological and biochemical analysis revealed that natural variations in amino acids can affect the degree of drug resistance (DR) conferred by some mutations. 48 It has been shown that HIV-2 and group O HIV-1 viruses are naturally resistant to nonnucleoside RT inhibitors (NNRTIs) due to mutations present in their RT gene. 49,50 Moreover, differences in nucleotide and mutational motifs (these are transitions and transversions needed to develop DR to different antivirals) between subtypes can affect the genetic barrier for resistance. 51,52
One good example of this is the V106M polymorphism in the RT of subtype C viruses inducing resistance to NNRTIs. 53 However, study of the influence of genetic variability and polymorphisms on HIV-1 DR development in places where diverse HIV-1 non-B subtypes, CRFs, and URFs are cocirculating is limited. We undertook this study in Abuja, Nigeria's capital city, using specimens collected from HIV-1-infected patients who were eligible for initiation of ART at two treatment sites. The aims of this analysis were to (1) determine the HIV-1 subtype distribution in the cohort; (2) identify and characterize baseline polymorphisms and DRMs at pretreatment and the association of any specific mutational pattern with HIV-1 subtypes or CRFs; and (3) evaluate the potential impact of these polymorphisms on DR development.
Materials and Methods
Specimens
Patients were recruited for a prospective cohort study to monitor HIVDR development. The median age was 34 years [interquartile range (IQR), 28–40 years]; 38% were male, while 62% were female at the time of ART initiation. The median CD4 count was 149 cells/μl (IQR, 73–205 cells/μl) and 72% of participants had a CD4 count of ≤200 cells/μl. Detailed demographic and clinical data of this cohort were described previously. 54 The specimens were collected from HIV-infected patients initiating ART from two sites in Abuja, between January and July 2008. Samples were systematically collected from all the patients who were eligible for ART to limit the introduction of bias in the nature and quantity of polymorphisms and/or DRMs detected.
RT-PCR and DNA sequencing
Sequences were generated from these plasma specimens using the broadly sensitive HIV-1 genotyping assay that amplifies the 5′ region of the HIV-1 pol gene including DR mutation sites in the PR and RT regions. 55 Sequences were edited with the ReCall program 56 and the consensus sequences were made for further analysis.
HIV-1 subtype determination and characterization
HIV-1 subtypes and CRFs for the newly obtained 271 sequences were initially determined by phylogenetic analysis using MEGA 557 along with 109 reference sequences including subtypes A to K and all available CRFs, downloaded from the Los Alamos HIV sequence database (2010 version,
The phylogeny of 84 subtype G sequences identified from the initial subtyping was further assessed by using the MrBayes tool (Geneious, v.6.1.8, Biomatters Ltd., San Francisco, CA) with the references of subtype G (n=36) and other major HIV-1 subtypes (n=21) downloaded from the Los Alamos HIV sequence database (
Identification of major drug resistance-associated mutations and natural polymorphisms
Major HIVDR mutations of the 271 quality-assured sequences were identified by the Calibrated Population Resistance Tool (CPR), version 6.0, deployed at the Stanford HIV drug resistance website (
Results
HIV-1 subtype distribution
Phylogenetic analyses of the 271 newly obtained nucleotide sequences along with 109 HIV-1 subtype and CRF reference sequences revealed that 93.7% (254/271) of the sequences could be subtyped with our sequence analysis procedures while 6.3% (17/271) could not be assigned a standard subtype or CRF, which were denoted as unclassifiable sequences (Table 1). A total of 16 possible types of HIV-1 strains were identified. The predominant strains were subtype G (31.0%), CRF02-AG (19.2%), and CRF43-02G (18.5%), accounting for 68.7% of the total sequences analyzed. The remaining strains (31.3%) were A/CRF36-cpx (11.4%), CRF06-cpx (5.5%), CRF25-cpx (3.3%), CRF36-cpx (2.2%), A/CRF02-AG (1.5%), CRF15-01B/G (0.4%), CRF43-02G/G (0.4%), and unclassifiable (6.3%). Overall, the recombinant viral strains, including CRFs and URFs, accounted for 68.6%, while pure subtypes (G and C) were only 31.4%.
CRF, circulating recombinant form.
The figures in bold are subtype or CRFs having higher prevalence.
It is interesting to note that the subtype G sequences identified in this study (n=84) exhibited intrasubtype diversity by segregating into two large distinct clusters in the initial phylogenetic analysis (data not shown). To further understand the diversity of these G sequences, we analyzed them with all available subtype G reference sequences from the Los Alamos database. Results indicated that the G sequences mainly formed three major and two minor clusters (Fig. 1). For description purpose, we named them G1-3 and Gm1-2, respectively. Most of the G sequences from this study (n=64) fell into two of the three known major subtype G clusters, G1 (n=19) and G3 (n=45), and only one clustered in the G2 along with reference sequences from other countries. Interestingly, the remaining study sequences formed two independent minor clusters of Gm1 and Gm2 without clustering with any sequence from other countries with an exception of two sequences that didn't group into clusters.
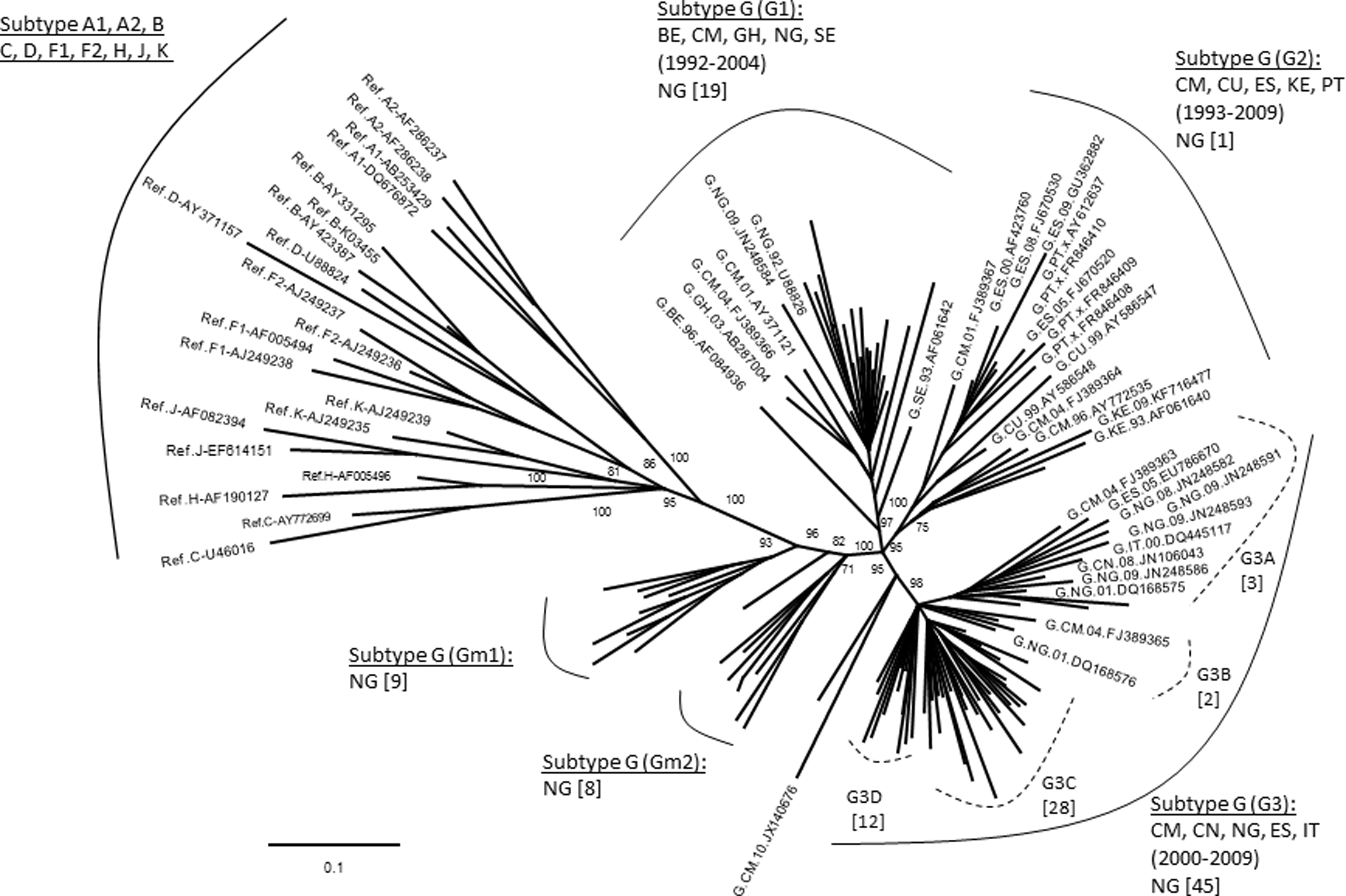
Phylogenetic analysis of subtype G sequences with HIV-1 reference sequences. The tree was constructed with 84 newly characterized Nigerian subtype G sequences and 57 reference sequences downloaded from the Los Alamos HIV database (
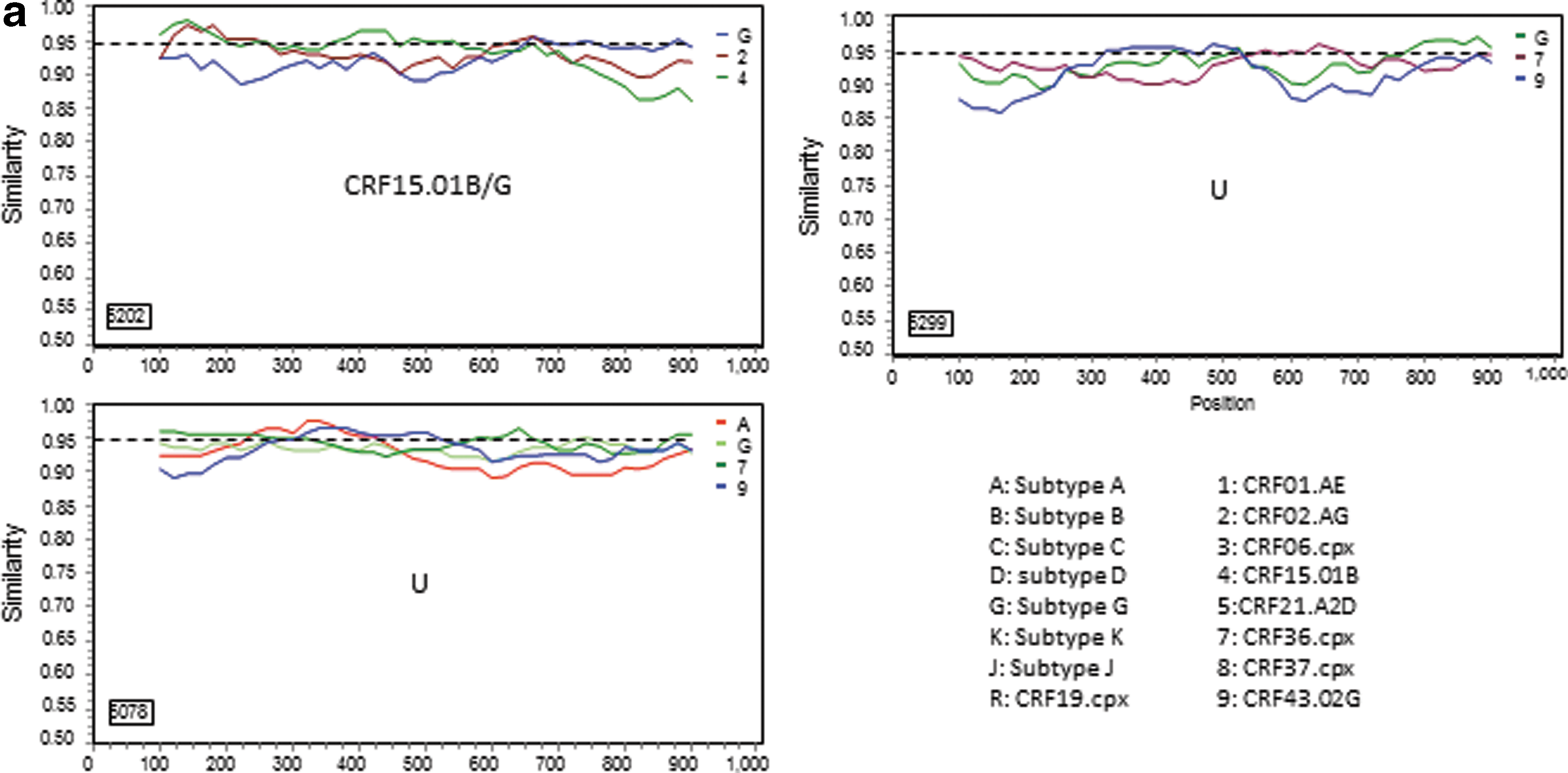
For those 17 sequences that a subtype/CRF could not be assigned, 13 representatives were selected for further analyses. Genetic distance analysis showed that they were not closely related to any subtype reference sequences, but had approximately equal distances to two or multiple subtype sequences (Table 2). Sequence similarity analysis indicated that they had complicated gene structures with multiple subtypes involved (Fig. 2a). Bootscan analysis further revealed that eight of them had strong evidence of recombination between multiple subtypes (Fig. 2b). Blast search for closely related sequences in the GenBank database showed the highest similarity hits were 93–95% to the GenBank sequences (data not shown), indicating they might be new recombinant viruses.
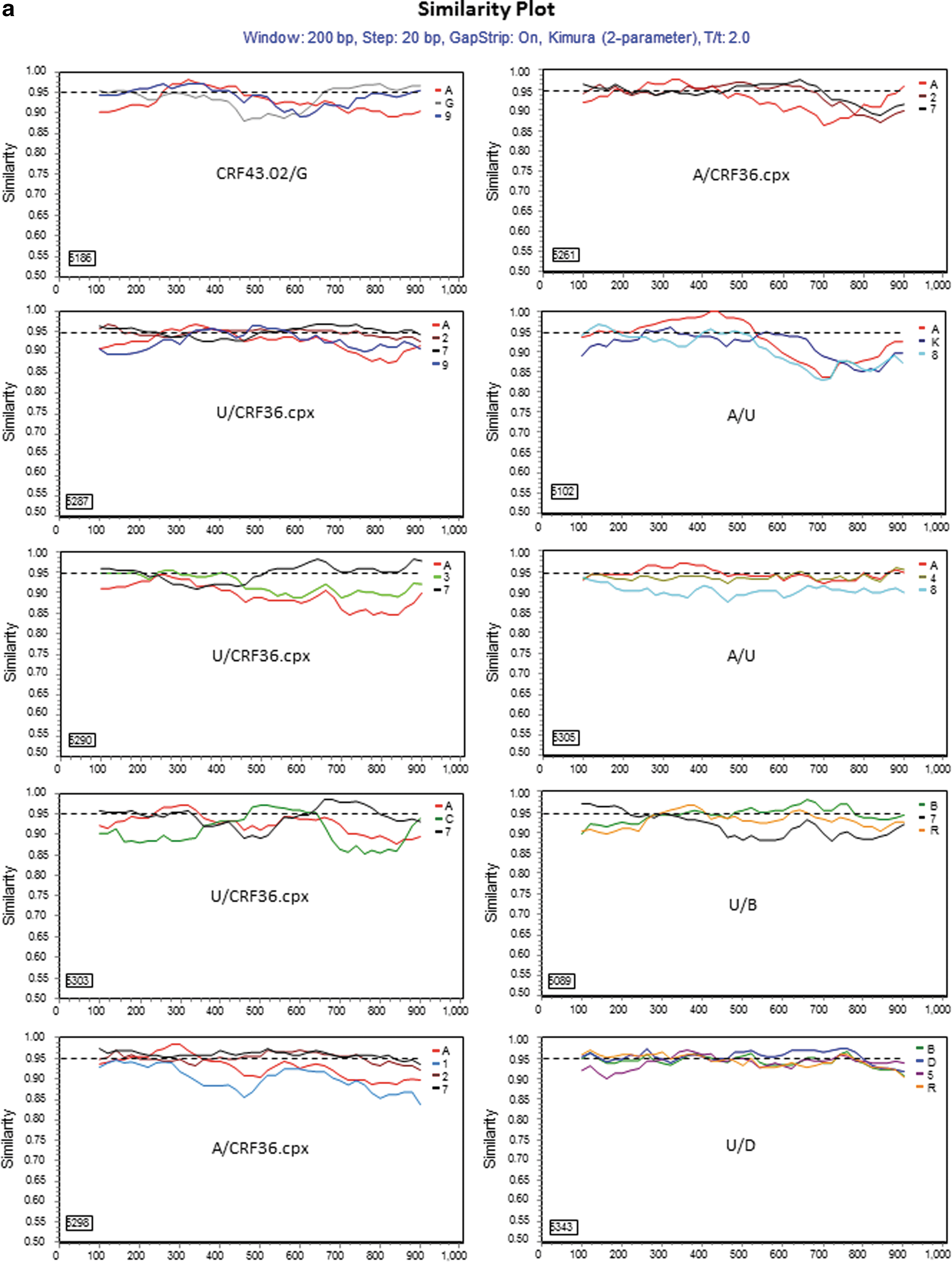
Further characterization of phylogenetically unresolved Nigerian sequences. The 13 representative Nigerian unclassifiable sequences (Table 1) were further analyzed individually using SimPlot
59
for genetic similarity (200-bp window, 20-bp step, and two-parameter Kimura algorithm). The selection of HIV-1 reference subtypes for analysis was based on the genetic distance analysis (Table 2). Only three to four relevant reference subtypes were chosen to generate the graph to show the gene structure of query sequences
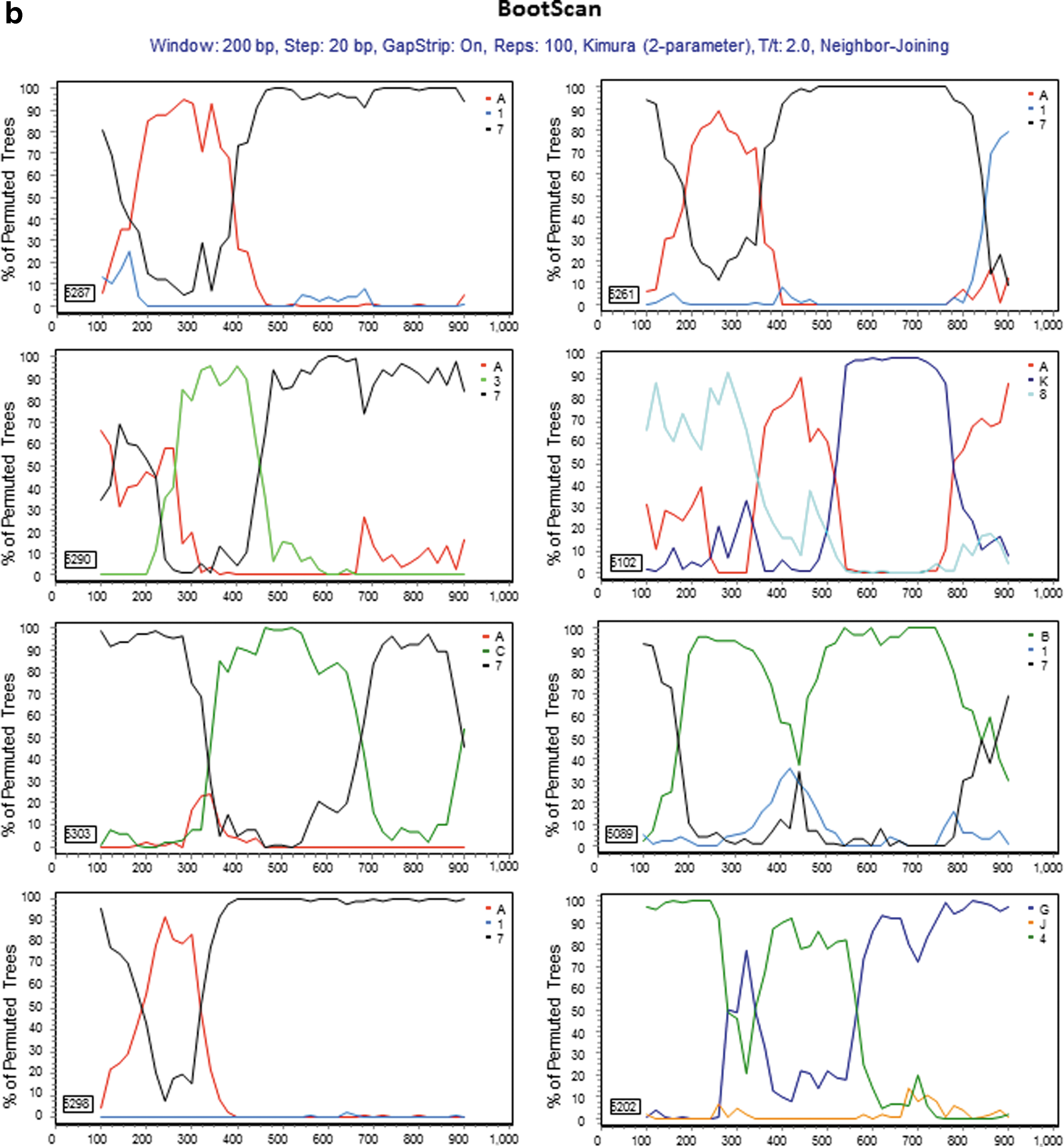
Details of methodology were described in Materials and Methods.
HIV-1 drug resistance-associated mutations
Among the 271 sequences analyzed, 28% (76/271) had polymorphisms and/or primary and/or accessory DRMs in the PR and RT region (Table 3). The CPR analysis identified the PI mutations M46L, I85V, and F53Y as well as the NRTI mutations M41L and V75M, and the NNRTI mutations K101E, K103N, and G190A as Surveillance Drug Resistance Mutations (SDRM). 61,62 The total number of detected mutations in the PR was 11, distributed at 9 sites among 11 samples. In the RT, 81 mutations were identified and distributed at 15 sites among 69 samples. Seven specimens had PI mutations but no NRTI or NNRTI mutations, while the remaining 69 specimens with resistance-associated mutations had no PI mutations.
Sensitivity column is filled only when drugs are resistant.
Potential low level of resistance is classified as sensitive as per WHO interpretation.
NFV, nelfinavir; EFV, efavirenz; NVP, nevirapine; ETR, etravirine; RPV, rilpivirine; AZT, zidovudine; d4T, stavudine; ABC, abacavir; 3TC, lamivudine; ddI, didanosine; FTC, emtricitabine; TDF, tenofovir.
In the PR region, one sample displayed the K43T mutation; another one had the M46L, while a third specimen had K53Y, A71V, and I85V. The T74S mutation was found in the PR of six specimens.
We also detected NRTI selected mutations M41L, E44D, T69ANS, V75M, and V118I, and NNRTI mutations V90I, A98G, K101EQ, K103NR, V106I, V108I, E138A, V179EI, and G190A (Table 3). Due to the M41L mutation detected in one specimen, the virus would have reduced susceptibility to zidovudine (AZT) and stavudine (d4T). Five specimens had A98G, K101EK, V108IV, K103N, E138A, and G190A occurring alone or in combination with each other, which could cause intermediate and/or high-level resistance to delavirdine (DLV), efavirenz (EFV), etravirine (ETR), and nevirapine (NVP). The mutation E138A associated with a decreased response to ETR, the second generation NNRTI, was found in 16% (12/76) of the specimens displaying DR mutations. There were only three specimens that had DR mutations associated with two combined drug classes (NRTIs plus NNRTIs). The overall resistance rate within the cohort was <3% and the affected drugs were one PI [nelfinavir, (NFV)], two NRTIs (AZT and d4T), and five NNRTIs [EFV, DLV, ETR, rilpivirine (RPV), and NVP].
Among all the mutations detected, T74S (54.5%) in PR and E138A (17%), V179I (16%), followed by V118I and V179E (14% each) in RT were the most prevalent, while T69NS, V90I, and V106I were present at only 5% each. Within the cohort, the rate of these mutations was 2.2% for T74S, 4% for V118I and V179E each, 4.5% for V179I, and 5% for E138A. The prevalence for T69NS, V90I, and V106I was 1.5%, respectively. L89T/I, the nonpolymorphic PI-selected mutation of uncertain phenotypic and clinical significance was also detected.
Natural polymorphisms
Of the 271 sequences analyzed, 11 polymorphic mutations (G16E, K20I, L23P, E35D, M36I, N37D/S/T, R57K, L63P, and V82I) in the PR and 3 (D123N and I135T/V) in the RT were identified. According to the Stanford DR algorithm, these substitutions occurred in the wild type (WT) of non-B subtype viruses and have no effect on DR for PIs or RTIs; however, when combined with other mutations, they increase the resistance to PIs or RTIs in subtype B viruses.
Uncommon mutations
Numerous uncommon mutations with unknown impact and/or function within the PR and the RT were identified. In PR, 58 uncommon mutations were detected at 28 positions, while in RT the number was 96 at 58 positions. Some of these mutations occurred at a very high rate and were subtype specific in the PR gene. For example, the E35Q mutation occurred in 67% of all subtype G sequences and in only 5% of CRF02-AG sequences, but was absent in all other subtypes and CRFs. E35K occurred in 5% of CRF02-AG but was absent in the rest of the other subtypes. However, in the RT gene, we did not detect a high prevalence of an uncommon mutation or subtype-specific mutational pattern. The random mutation rates ranged from 0.5% to 15%.
Synonymy and subtype-specific codon mutational pattern
We next performed a codon-by-codon comparison of the nucleotide sequences of subtype B (HXB2) to the sequences in the cohort to detect any subtype-specific mutational patterns (Table 4). We found that at position 179 of the RT gene, all sequences of CRF06-cpx (6/6), subtype G (2/2), and CRF43-02G (2/2) harbored the V179E mutation, while all sequences of CRF36-cpx (1/1), A/CRF02-AG (2/2), CRF36-cpx/B (2/2), A/CRF15-01B (4/4), and CRF25-cpx (2/2) had the V179I mutation. The WT CRF06-cpx and subtype G mainly had a codon GTG at position 179, while the WT of the other subtypes and CRFs possessed the GTT at this position except for CRF36-cpx, which had a codon ATA.
Discussion
We have analyzed the PR and RT regions of the pol gene of 271 sequences generated from plasma specimens collected from ART-naive HIV-1-infected patients initiating ART in Abuja, Nigeria in 2008. Phylogenetic analysis revealed that recombinant viruses have become the dominant strains over pure subtypes. The subtype G viruses have further diversified into three major subtype G clusters using worldwide sequences, and most Nigerian sequences from this study were closely related to two of the three clusters (Fig. 1, G1 and G3), indicating a close epidemiological relationship of HIV epidemics between Nigeria and countries in Europe, West Africa, and Asia during the periods of 1992–2004 (G1) and 2000–2009 (G3), respectively. We also noticed that the HIV-1 strains in the G2 cluster formed by sequences obtained from countries in Europe, East and West Africa, and South America from 1993 to 2009 had a minimal impact to the HIV epidemic in Nigeria, and strains in the two minor clusters, Gm1 and Gm2, appear to be indigenous viruses that had no relationship to the HIV subtype G strains circulating in other countries.
The three major subtype G clusters were reported previously as GWA-I, GWA-II, and GCA. 6,22,63 However, within a G cluster, different strains may be present. For example, in the G3 cluster, there are possibly four different strains, denoted as G3A–D (Fig. 1), while strain G3C and G3D seem to circulate only in Nigeria. Studies have suggested that subtype G emerged in Central Africa in 1968 (1956–1976), and between the middle and late 1970s two subtype G strains were probably introduced into Nigeria. 63 Since then, they were disseminated locally and to neighboring countries, leading to the origin of two major western African clades (GWA-I and GWA-II). For the strains in the two minor clusters of Gm1 and Gm2, we have not identified any phylogenetic relationship of these strains to those from other countries (>95% similarity) or to those detected in other regions of the country (data not shown), indicating that they might be newly emerging and locally circulating only in the Abuja area.
We also observed an evolved subtype distribution in the country. Prior to 2000, subtypes A (61%) and G (31%) were dominant in the south and north, respectively, and CRF02-AG was not detected in the country. 18 However, after 2000, studies revealed that the prevalence of subtype A was greatly reduced with a slight reduction of subtype G and a remarkable increase of CRF02-AG (39–57%). 16,34,38 –41 In this study, we did not detect pure subtype A and had rates for subtype G (31.0%) and CRF02-AG (19.2%) detected in the capital area of Abuja similar to those detected in other areas, which is in agreement with the subtype distribution patterns reported in the country. 16,34,38,39 Our data and other studies suggest that among the two previously dominant subtypes, one (subtype A) is disappearing and the other (subtype G) is still playing an important role but is circulating at different rates in the country. Meanwhile the progeny (CRF02-AG) of subtypes A and G are emerging quickly in the HIV epidemic in the country. In addition, CRF43-02G is becoming one of the dominant subtypes in Abuja, which was not reported in other areas of the country. Nevertheless, the majority of the current circulating HIV strains were derived from the subtypes or recombinant forms between subtypes A and G, again indicating localized transmission and circulation.
The overall pretreatment HIVDR rate in the cohort was less than 3%, which is expected due to the short time of ART implementation in the country at the time of the study. This rate is in agreement with the rates reported in Jos, in the Plateau State, 40 and the North Central region of Nigeria. In the protease region, one sample displayed the K43T mutation, which was associated with a decreased virological response to tipranavir boosted with ritonavir (TPV/r) in the RESIST trials. 64 One sample harbored the M46L mutation, which is a primary mutation inducing an intermediate level of resistance to NFV. A rare treatment-associated mutation K53Y, the polymorphic mutation A71V, as well as the PI-selected mutation I85V were also found in one sample each. Six specimens had the T74S mutation that induces a potential low level of resistance [classified as sensitive per the World Health Organization (WHO) interpretation] 65 to PIs.
We also identified subtype/CRF-specific mutation patterns in the PR and RT gene regions. For instance, K20I in the PR region was present in all subtypes and CRFs except for eight sequences that were all subtype A-containing recombinants. 34 The polymorphic mutations M36I, L63P, V82I, and M89I were proportionately present in the sequences of CRF02-AG, CRF43-02G, and CRF06-cpx and subtype G. A study has shown that the polymorphic mutation M36I in the PR region was associated with a higher rate of PI treatment failure. 66 Furthermore, biochemical studies of this mutation with subtypes A and C recombinant I36 PR revealed that the PR enzymes of non-subtype B viruses have a higher inhibition constant (K i) and a greater catalytic efficiency than subtype B viruses 67 and that introduction of I36 into subtype B HIV-1 resulted in a higher virus replication capacity in both the absence and presence of PIs. 7 In addition, we found that mutations E35D, N37DST, R57K, and K70R in the PR and D123N and I135TV in the RT were present at high rates in this cohort. These mutations have been shown to be associated with increased or decreased DR to some ARVs. 68 –70 Several other mutations observed in this study were not classically associated with resistance and their biological functionality remains to be elucidated. Since some of the uncommon mutations occurred at a very high frequency in a subtype/CRF-specific manner, they may warrant further investigation to determine their biological functions within the virus genome.
The analysis of the mutational pattern suggests that the V179I mutation in the RT is not only the preferred mutation in subtype B viruses, but also occurs in non-B subtypes and CRFs. Subtype G, CRF43-02G, and CRF06-cpx viruses utilized the codon GTG/GTA to encode valine and more likely to develop V179E mutations, 16,34,38,39,71 whereas other CRF viruses are biased toward V179I and V179D. This pattern of amino acid substitution by a single nucleotide change is called quasisynonymy; it may explain the high rate of the V179E found in subtype G and CRF06-cpx in this cohort and is concordant with published data and statistics predicting the occurrence of this mutation at RT position 179 of these viruses. 16,71,72
In summary, this study has shown an evolving HIV-1 epidemic in Abuja, with recombinant viruses becoming the prevailing strains and locally circulating strains emerging. This evolving trend in the HIV-1 epidemic deserves closer monitoring since a recent study has shown that a recombinant HIV-1 sub-subtype A3 and CRF02-AG virus was more virulent than the original viral strains. 73 In addition, it has also confirmed that the occurrence of DR may be influenced by quasisynonymy and the genetic cost of mutations.
Sequence Data
The GenBank accession numbers for sequences analyzed in this study are JX083986–JX084256.
Footnotes
Acknowledgments
This research was supported by the President's Emergency Plan for AIDS Relief (PEPFAR) through the Centers for Disease Control and Prevention.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention/the Agency for Toxic Substances and Disease Registry.
Author Disclosure Statement
No competing financial interests exist.