
Abstract
The current antiretroviral therapy (ART) has suppressed viremia to below the limit of detection of clinical viral load assays; however, it cannot eliminate viremia completely in the body even after prolonged treatment. Plasma HIV-1 loads persist at extremely low levels below the clinical detection limit. This low-level viremia (termed “residual viremia”) cannot be abolished in most patients, even after the addition of a new class of drug, i.e., viral integrase inhibitor, to the combined antiretroviral regimens. Neither the cellular source nor the clinical significance of this residual viremia in patients on ART remains fully clear at present. Since residual plasma viruses generally do not evolve with time in the presence of effective ART, one prediction is that these viruses are persistently released at low levels from one or more stable but yet unknown HIV-1 reservoirs in the body during therapy. This review attempts to emphasize the source of residual viremia as another important reservoir (namely, “active reservoir”) distinct from the well-known latent HIV-1 reservoir in the body, and why its elimination should be a priority in the effort for HIV-1 eradication.
Introduction
C
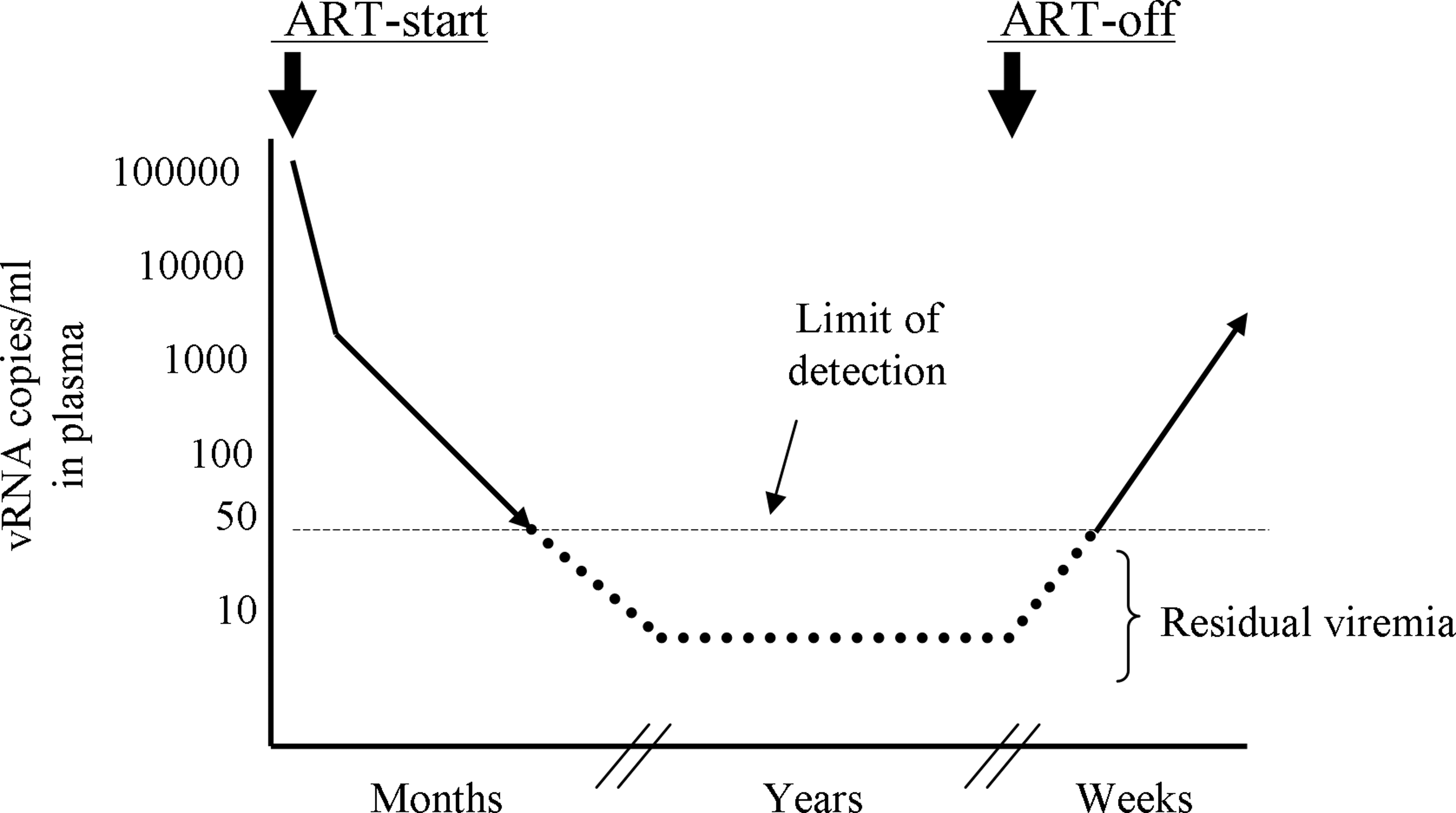
Antiretroviral therapy (ART)-mediated suppression of viral loads in patients. Residual viremia persists below the clinical detection limit during prolonged ART. Viral loads return to even pretreatment levels within weeks of therapy withdrawal.
Over the years, free virus in plasma of patients on suppressive therapy is frequently detected below the clinical detection limit by using ultrasensitive polymerase chain reaction (PCR) techniques that amplify viral sequences in samples with as low as a single copy target. 10 –15 This low-level viremia is often termed “residual viremia.” The single copy vRNA detection assay can detect ranges of vRNA from 1 to 32 copies in plasma of patients with effective virus suppression by ART. 9,16 The clinical significance of residual viremia is still not clear, nor is its cellular source in patients on therapy. The following sections briefly highlight the attempts that have been made to characterize residual viremia as well as to uncover its clinical importance during prolonged ART.
Residual Viremia Still Persists After Treatment Intensification
One explanation is that residual free viruses in plasma are produced due to incomplete inhibition of virus replication by ART in the body, especially in the lymphatic tissues where suboptimal antiviral concentrations persist. 17 Viral replication is referred to here as the continuous cycles of viral infection, integration, and production from target cells. The incomplete viral inhibition is supported by some studies that found increased levels of HIV-LTR circles in patients' peripheral blood mononuclear cells (PBMCs) after therapy intensification with an integrase inhibitor, raltegravir. 18,19 In addition, if excessive amounts of virus particles are available for target cell infection in tissues during cell-to-cell virus transmission, there is a probability that the virus might escape from optimal drug inhibition in infected cells, 20 leading to viral DNA (vDNA) integration and production of viral progenies. In such a scenario, therapy intensification with the integrase inhibitor could also result in further inhibition of low-level viral replication, reflecting the increased levels of HIV-LTR circles in PBMCs.
However, the treatment intensification with additional antiretrovirals added to the drug combination could not significantly reduce the levels of residual viremia in patients. 19,21 –26 If the persistence of residual viremia consisting of short-lived free viruses 3 –6 is the result of continuous cycles of virus replication occurring in tissues during ART, then the evolution of drug-resistant residual viruses would be prevalent, which is generally not found in patients with suppressive therapy. 10,27,28 Moreover, there is evidence demonstrating that virus replication is almost completely blocked in tissues during ART. 29 –31 These data point to a scenario in which under optimal antiviral concentrations, residual viremia might arise as a result of persistent release of the virus at low levels from the highly stable productively infected cells. 25,32,33 To persist in the body, these infected cells must have the ability to survive from both viral cytopathicity and immune-mediated clearance. Such persistently virus-expressing cells, if they exist in the body, should represent the “active HIV-1 reservoirs,” while in contrast the latent HIV-1 reservoirs (discussed below) carry transcriptionally silent viruses requiring stimulation to enter their productive phases.
Unknown Source of Residual Viruses in the Body
HIV-1 can persist as latent but inducible forms in resting memory CD4 T cells in patients, even during prolonged ART. 34 –36 The frequency of latently infected cells is about 1 per 106 resting CD4 T cells. 37,38 These cells remain extremely stable in the body with an estimated half-life of about 44 months, 39 and can produce the virus when reactivated. Essentially, latently infected cells are maintained in the body for the lifetime of treated patients, 39 accounting for an important HIV-1 reservoir in the body. 40 This latent reservoir has been a major obstacle to virus eradication by using ART. 39 The initial suggestion was that the intermittent reactivation of latently infected resting CD4 T cells in vivo results in persistent residual viremia during therapy. 10,28 In support of this notion, occasionally, some studies have reported the close genetic relationships between latent virus residing in resting CD4 T cells and residual cell-free virus present in plasma below the clinical detection limit during ART. 27,41
However, two studies reported in 2009 compared residual plasma viral sequences with blood CD4 T cell-derived viral sequences in a number of patients on effective ART using phylogenetic and sequence compartmentalization tests. 42,43 In these studies, residual plasma viral sequences appeared more genetically homogeneous than their CD4 T cell-derived counterparts. The majority of plasma viral sequences were even clonal in some patients. 43,44 The notion that the intermittent reactivation of highly heterogeneous latent virus population present in blood CD4 T cells leads to residual viremia was not supported by these observations. Furthermore, viral sequences obtained from these different sources (i.e., plasma and CD4 T cells) were found to be compartmentalized separately. 42,43 These data led to the suggestion that the majority of residual plasma viruses did not originate from the blood CD4 T cell compartment in patients on suppressive therapy; rather these data indicated the existence of a unique but yet unknown source(s) of residual plasma viruses in the body. 42,43 If true, the source of residual virus should represent an “active” reservoir for HIV-1, which remains highly stable in the body and perhaps persistently releases the virus at low levels during suppressive ART. 33
The characteristics of unknown cell type(s) harboring residual viruses are not clear, but based on viral characteristics several features could be predicted (see Table 1). The analyses of residual viral env sequences in a number of patients suggested that these viruses use either CCR5 or CXCR4 as the coreceptor during infection of target cells. 44 –46 This indicates that the residual virus's source may express both CCR5 and CXCR4 molecules on the cell surface. A low proportion of residual viral tat sequences isolated from some patients possessed missense mutations, resulting in the loss of function of viral Tat. 42,46 The detection of defective tat mutants in plasma during prolonged effective therapy suggests that residual viruses are produced in cell types that are capable of synthesizing viral mRNA without viral Tat, which is generally required for efficient viral transcription from LTR. This is not surprising because there are reports showing that some infected cells stimulated with tumor necrosis factor (TNF)-α can support the replication of tat-defective virus. 47 This, in turn, perhaps provides a clue for the possible sites of residual virus production in the body. For example, at the inflammatory locations in the body, residual virus-infected cells with elevated levels of active NF-κB due to cytokine signaling may persistently produce virus at low levels even during ART.
Because of the apparent clonality of residual viruses in some patients, the cell source of these viruses is thought to have a proliferative capacity for which identical copies of these viruses are made. 44 One such source is predicted to be the hematopoietic stem/progenitor cells of monocyte–macrophage lineage, 44 which still remains to be proven. Although HIV-1 infection in hematopoietic CD34+ stem cells remains controversial, 48 –52 recent data suggest that a proportion of these cells can be infected with laboratory-adapted HIV strains. 53 –55 Yet the isolation of HIV sequences directly from patients' blood-derived CD34+ cells has not been widely reported, 56,57 although any negative results would also point to the scarcity of HIV+CD34+ cells in patients' blood. Of note, the HIV infection in CD34+ cells might not necessarily lead to the incorporation of proviral DNA in multiple hematopoietic lineages in vivo. This is because reports have shown that HIV-1 Nef can impair the differentiation of an early T/NK cell precursor and the development of T cells in vitro. 58 –60
Recently, there have been some interesting developments in the field of the HIV-1 reservoir. It has been discovered that a small proportion of T cells, including CD4+ T cells, in humans 61 and also animals 62 possesses stem cell-like properties, such as the capacity to self-renew and differentiate into effector memory, central memory, and terminally differentiated effector T cell subsets. These cells are termed “stem cell-like memory T cells” (TSCM). In 2014, Buzon et al. 63 showed that CD4+ TSCM are susceptible to HIV-1 infection in vitro, and detected vDNA in a portion of these cells isolated from several patients on prolonged ART. In addition, they recovered replication-competent virus in viral outgrowth assays from three treated patients. 63 These data suggested that CD4+ TSCM cells also serve as an HIV-1 reservoir in vivo, like other memory CD4 T cell subsets. 34,64,65 In the same year, two studies 66,67 analyzed proviral DNA integration sites in CD4 T cells isolated from patients receiving prolonged suppressive therapy, and discovered that a large proportion of integrations occurred in the host genes involved in the regulation of cell proliferation and survival, and also in the development of cancer. In essence, these data revealed the clonal expansion of some CD4 T cells with integrated vDNA in vivo during long-term suppressive therapy, similar to the observations made previously. 68 –70 However, these studies provided a potential mechanistic explanation about how a population of CD4 T cells with integrated vDNA could undergo clonal expansion in vivo. 66,67
Whether these clonally expanded infected cells possess replication-competent vDNA or continuously release virus at low levels resulting in residual viremia during ART is not clear. However, interestingly among these studies, Maldarelli et al. 66 found that the plasma-derived clonal population of viral gag-pro-pol sequences exactly matched the sequence obtained from a clone of expanded CD4 T cells in a patient on ART. The proposition is that the clonally expanded CD4 T cell population with proviral DNA might be a source of residual viremia in this patient during therapy. Although this observation is encouraging, such viral sequence comparisons need to be performed in a number of patients to confirm the likelihood of these clonally expanded cells being the source of residual viremia during suppressive therapy. Nonetheless, a caveat will exist—for example, if patients miss doses of antiretrovirals inadvertently, even without having a measurable blip in viral loads, there is a possibility that replication-competent residual plasma viruses 46 originating from an unknown source may enter the CD4 T cell compartment and persist long term. In such a scenario, these viruses will be underrepresented in the CD4 T cell reservoir, as often observed. 42,43 However, the rare detection of these viruses in this reservoir using sequence comparison tests may mistakenly lead to the conclusion that the CD4 T cells are the source of residual viremia during therapy. Eliminating a potential source for the virus by an eradication strategy with the simultaneous decay in residual viremia would conclusively establish the link between the targeted viral source and residual viremia in patients on therapy.
Unlike productively HIV-infected CD4 T cells (t 1/2 ∼1 day), infected macrophages (MΦ) are relatively resistant (t 1/2 ∼2 weeks) to virus-induced cytopathicity. 71,72 Therefore, HIV+ MΦ in various tissues, e.g., in gut and central nervous system (CNS), may also potentially represent the crucial source of residual HIV-1 during ART. 73 –75 The possible contributions of a small proportion of other cells, such as astrocytes 76 –82 or microglia, that are found to be infected in the body cannot be ruled out as the source of residual viruses. Essentially, any virus-infected cells present in the body capable of producing virus particles somewhat persistently could potentially be a reservoir for residual plasma viruses during therapy.
Formation of Residual HIV-1 Reservoir: A Hypothetical View
Since residual plasma viral sequences and blood CD4 T cell-derived viral sequences are separately compartmentalized in some patients on ART, 42,43 at least two hypotheses can be formulated: (1) residual viruses are produced in some infected CD4 T cells in the body that do not usually circulate in patient's blood; and (2) residual viruses might be produced in non-CD4 T cell reservoirs. However, how the residual virus reservoir is formed in patients is yet to be defined. One prediction might be that prior to ART initiation, a minor population of HIV-1 variants representing residual viruses among the viral quasispecies in the body (Fig. 2) might have been tropic to one or more unknown cell types for infection, which are naturally stable in the body. However, these infected cell types must have avoided virus-specific immune responses and viral cytopathicity for their long-term survival as well as for the persistent release of viruses in the body. Whereas viral loads quickly decline after ART initiation 1,2 and eventually become undetectable after several months of treatment, 2 residual virus-infected cells still continue to produce the virus at low but perhaps fluctuating levels, 83,84 giving rise to residual viremia during suppressive ART.
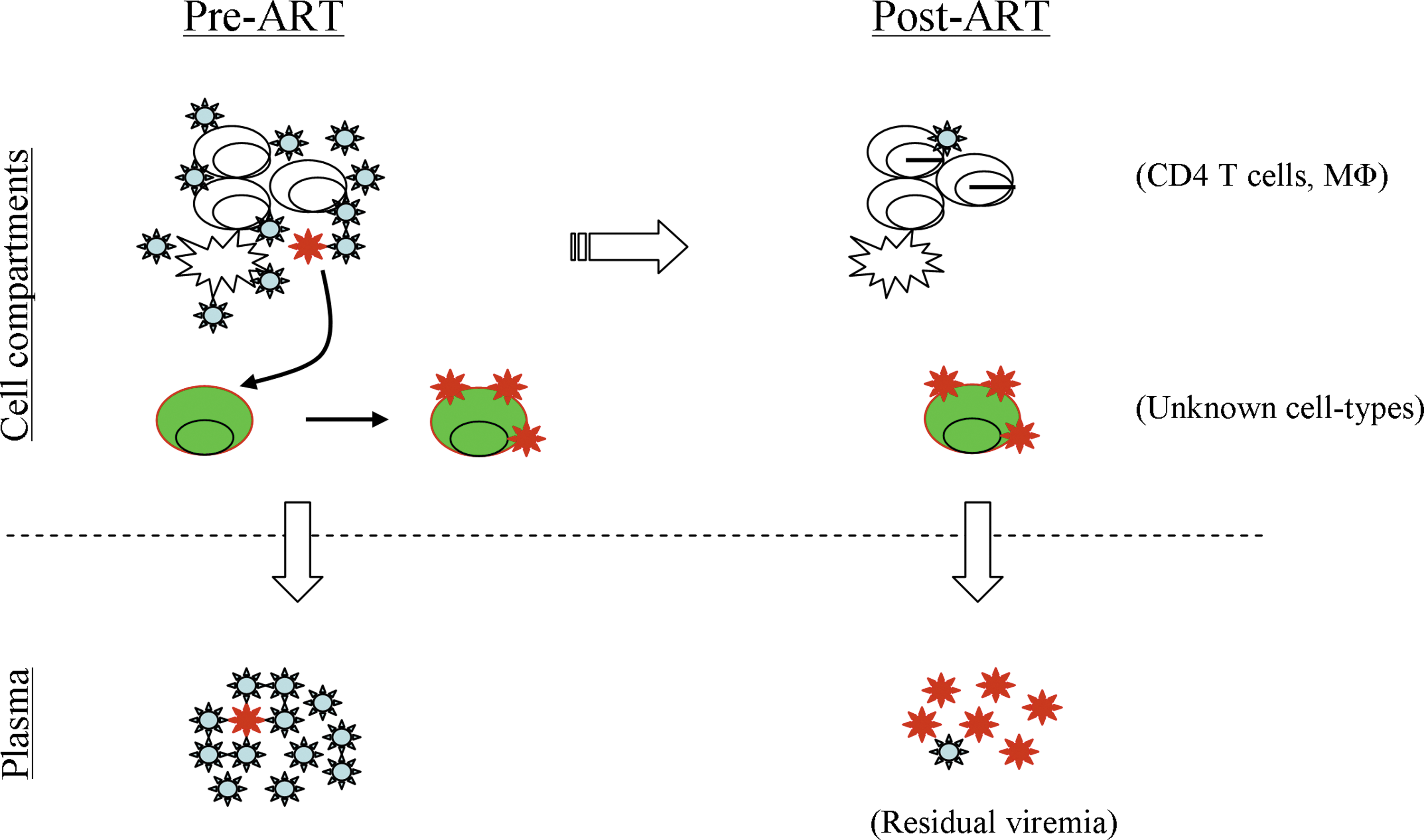
A conceptual view for the establishment of residual virus's source. Prior to ART (left), most plasma viruses (lower left) are produced by infected CD4 T cells and macrophages, but a minor population of virus (in red) among viral quasispecies (upper left) can become tropic to a yet unknown cell type(s) (in green) and establish the residual virus reservoir. During prolonged ART (right), highly stable, infected green cells persistently produce virus at low levels, contributing most to residual viremia (lower right), whereas CD4 T cells with reactivated HIV-1 from latency might be the minor contributors to this low-level viremia. Color images available online at
In support of the above prediction, Kearney et al. 29 have shown that in the absence of virus replication and evolution in patients on prolonged effective therapy, the persistent residual plasma viruses with identical sequences appear to be released from a clonally expanded stable cell population that was likely infected prior to therapy initiation. Although the nature and the identity of this cell population are yet to be uncovered, other studies have found evidence for clonal expansion of some HIV-infected CD4 T cells in patients on prolonged therapy, as mentioned before. 66,67 Whether residual plasma viruses originated from clonally proliferated CD4 T cells in the majority of patients on therapy is not clear. However, it becomes gradually obvious that the underlying mechanisms of how the reservoir for residual virus is established in vivo prior to therapy and how the virus is released persistently from infected cells during therapy would remain important for future investigations.
Replication Potential of Residual Plasma Viruses
Although low-level viremia is frequently detected in patients on effective ART below the detection limit of clinical viral load assays, it is not fully clear whether residual vRNA represents genetic material of replication-competent virus particles in the body. Because of error-prone replication of HIV-1, 85 these residual vRNA can easily be assumed to be part of replication-defective viruses. These defective viruses could be persistently released from the yet unknown productively infected cells in the body under ART. However, using residual vRNA isolated from a patient's plasma during ART-mediated viral load suppression to less than 75 copies/ml, residual viruses were fully reconstructed molecularly. 46 About half of these reconstructed viral clones were found to be replication competent, whereas the other half possessed missense and nonsense mutations known to cause defects in viral replication. 46 Nevertheless, the detection of replication-competent viruses represented in residual viremia of a patient warrants similar analyses in other patients on ART for verification purposes. If residual viruses in most patients are found to be replication competent, then these cell-free viruses circulating in plasma at low levels could pose as significant players in the evolution of drug resistance during low adherence to therapy, as well as in viral load rebound after therapy interruption (discussed below).
Possible Clinical Impact of Residual Viremia
The clinical implications of residual viremia are still not fully clear, but its occurrence could reflect various clinical scenarios as follows: chronic immune activation (CIA) is a hallmark of HIV-1 infection 86,87 and is associated with increased morbidity and mortality of infected individuals. 88,89 CIA is indicated by the elevated levels of proinflammatory cytokines in blood, such as interleukin (IL)-6, TNF-α, and interferon (IFN)-α and the higher percentages of circulating CD38+ T cells in patients than in normal individuals. 90 –93 At the early stage of HIV-1 infection, severe depletion of CD4 T cells occurs in gut-associated lymphoid tissue (GALT), 94 –96 and in the process, gut epithelial integrity is compromised, resulting in the leakage of gut microbes or microbial products to the bloodstream (termed microbial translocation). 97 –101 Microbial products, such as lipopolysaccharides (LPS), lipopeptides, and DNA containing CpG-motifs can activate immune cells, such as monocytes, dendritic cells, and others, through Toll-like receptors and induce secretion of various proinflammatory cytokines from these cells. 86,87 The recent experiments in SIV-infected pigtailed macaques suggest that the extent of immune activation is strongly associated with the rate of microbial translocation occurring in the gut. 102
Although ART can suppress viral loads effectively to below the limit of clinical detection assays, microbial translocation is not completely abolished in all patients 103,104 and CIA still persists during suppressive therapy. 45,105 –108 In elite controllers who naturally suppress viral loads to <50 copies per ml without therapy, higher percentages of CD38+HLA-DR+ T cells are also found to circulate in blood, compared to normal uninfected controls, 109 indicating that the persistence of low-level immune activation also exists in these natural viral load controllers. In fact, residual viremia persists below 50 copies per ml in plasma of these elite controllers. 110
Whether residual viruses play any direct role in causing CIA that persists during ART is not fully clear, but this possibility cannot be ruled out. 111 This is because the persistent production of HIV-1 particles (being infectious or not) below 50 copies/ml should constantly supply viral antigens at low-levels, which should result in some immunological consequences, such as the stimulation of plasmacytoid dendritic cells 112 –114 and monocytes 115 in the body. The proinflammatory cytokines produced as a result of such immune reactions at limited levels may partly contribute to the persistent immune activation observed in patients on effective therapy or in elite controllers. 116 This scenario is supported by the recent study by Hatano et al. in 16 asymptomatic HIV-1 controllers with median viral loads of 77 copies/ml. 117 These patients were treated with an ART regimen containing raltegravir for 24 weeks. The authors found that there was a trend toward a decrease in immune activation with a concomitant but significant reduction in viral loads in these patients after treatment. 117
Although the sample size was too small to reach a definitive conclusion in this study, the data suggested a causal link between low-level viremia and persistent immune activation in these asymptomatic HIV-1 controllers. 117 Similarly, in some ART-treated chronic HIV-1 progressors, the reduction in immune activation was also observed after treatment intensification with a raltegravir-added combined regimen. 19,23,118,119 This suggests that further suppression of “cryptic” HIV-1 replication in some tissue locations by therapy intensification (having no effect on residual viremia) leads to the attenuation of immune activation still persisting in treated patients. These observations collectively support the idea that the elimination of residual viremia might significantly reduce the levels of CIA present in patients on suppressive therapy.
Evolution of drug resistance
The current ART can effectively block virus replication in the body by hindering productive infection in the new target cells. 1,2 However, this therapy cannot inhibit the production of virus particles from previously infected cells with integrated proviral DNA. 120 In patients with low adherence to therapy, incomplete suppression of viral replication occurs due to suboptimal antiviral drug concentrations in the body. This increases the chance for the development of antiretroviral drug-resistant viral variants as a result of error-prone viral cDNA synthesis in the newly infected cells. 121 –123 If the newly released residual plasma viruses are proven to become replication competent in the majority of patients after ART interruption, 46 then these viruses should be the first to initiate infection and replication cycles in the body during suboptimal therapy adherence. This scenario is expected to accelerate the evolution of drug resistance in patients with fluctuating levels of adherence to therapy.
Viral load rebound after ART-off
ART suppresses viral loads to clinically undetectable levels, but in almost all patients viral loads return to pretreatment levels within several weeks after therapy interruption 124 –127 (Fig. 1). There is a degree of uncertainty about the source of viral load rebound soon after therapy interruption. 128,129 Generally, much of these rebounding viral loads are attributed to the latent HIV-1 reservoir in patients, 130 which is represented by the resting memory CD4 T cells with transcriptionally silent but inducible integrated proviral DNA. 64,131 The latent HIV-1 reactivation in resting memory CD4 T cells followed by virus spread in the body should theoretically result in the quick rise of viral loads after therapy interruption. However, a definitive proof for this is yet to come.
Alternatively, if residual viremia is the result of constant release of virus particles from one or more unknown cell sources, 25,32,33 and at least a portion of them remains replication competent, 46 then these viruses are expected to spread in the body as soon as antiretroviral drug pressure stops. Therefore, residual viruses might greatly contribute to early viral load rebounds after therapy is stopped. In support of this possibility, interestingly a study showed that the rebounding viruses in plasma of a patient 3 weeks after therapy interruption were phylogenetically much closer to residual plasma viruses present at the time of therapy interruption than the viruses of CD4 T cells. 42 Such analyses were possible because there was a measurable genetic distance between residual free plasma viruses and latent viruses of CD4 T cells present in this patient during effective therapy, 42 which is required for pinpointing the likely source of early rebounding viremia after therapy interruption in patients through viral genetic assessments.
Late rebound of viral loads in the “Boston patients”
The earlier case of Timothy Brown, the “Berlin patient” 132,133 who had shown prolonged remission of HIV-1, has energized the whole field to attempt to achieve a cure for HIV using new approaches. In 2007, Brown received an allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia after myeloablative conditioning. 133 Transplanted stem cells were obtained from a donor homozygous for the CCR5Δ32 mutation. 132 Since then, he has not been treated with ART, but posttransplantation tissue analyses for HIV-1 DNA and RNA suggest that he has been HIV free for more than 7 years. Clearly, viral reservoirs were either completely eliminated from his body or are present at extremely low but undetectable levels in an “inactive” status.
In contrast, two HIV-infected patients in Boston received allogeneic wild-type CCR5+ hematopoietic stem cell transplants for their hematological disorders after they were treated with reduced-intensity conditioning chemotherapy. 134 Initially during the posttransplantation period while they were on ART, viral nucleic acids could be detected in their blood CD4 T cells or plasma for some time (∼2–3 months), 134 but later these could not be detected for up to 4.3 years for one patient and 2.6 years for another in sensitive quantitative PCR assays. 135 Subsequently, these patients underwent analytical ART interruption when their blood and rectal mucosa both tested negative for HIV DNA and RNA. Despite a minimum of a three log10 reduction in reservoir size, both patients experienced viral load rebound—one patient at about 12 weeks and another at 32 weeks after ART interruption. 135 These data suggest that HIV remained hidden in long-lived reservoirs in their bodies at low amounts, but were enough for its slow spread on occasion and eventual burst of infection, giving rise to detectable viremia during an ART-free period. These reservoirs are likely the latent reservoirs that remain as an indisputably major obstacle for HIV eradication.
However, it is not clear whether the observed delays in viral load rebound after ART interruption is due to the substantial reduction in overall viral reservoir size, which is usually considered, 136 or due to the concurrent elimination of particular reservoirs, such as the “active” reservoirs in the body. An intriguing possibility exists that the “active” HIV reservoirs might have been eliminated somehow due to allogeneic hematopoietic stem cell transplantation, resulting in the delayed return of viral loads in these patients. Otherwise, these reservoirs could spread infection quickly and rebound viral loads much earlier after ART interruption than observed.
Viral load rebound in the “Mississippi baby”
An infant girl, who was born in Mississippi to an untreated HIV-infected mother, was treated with ART at 30 h after birth and the therapy was continued for the next 18 months. 137 After this period, the family stopped treating her with ART. At 24 and 26 months, multiple tests were performed on her blood samples to detect the presence of HIV. Although she was negative for HIV in most assays, one out of three wells with monocyte-derived adherent cells at 24 months and one out of six wells with PBMCs at 26 months tested positive for vDNA in PCR assays. 137 Interestingly, at 24 months, she also had residual viremia at a level of one vRNA copy per ml of plasma when three out of three wells with plasma tested positive for vRNA in a single copy viral load assay. 137 The child was declared negative for HIV at 26 months, although her blood cells possessed vDNA at a frequency of 4.2 copies per 106 PBMCs at this time. 137
Although replication-competent virus could not be recovered from her blood CD4 T cells, the reported assay results 137 could not fully rule out the possibility that she had extremely low levels of virus in her body. However, she remained untreated until the age of 46 months when her plasma viral load returned to clinically detectable levels. Clearly, the virus persisted in her body, presumably in latently infected resting CD4 T cells, or other cells at extremely low frequencies, which ultimately led to a rebound in viral load after a long absence of detectable viremia during the ART-free period. Based on our hypothetical model of “active” reservoir formation (Fig. 2), our speculation is that due to the early arrest of virus replication in the child by ART, the virus could not diversify itself enough to establish a residual virus reservoir (Fig. 2) in her body. The absence of such a reservoir capable of releasing virus persistently in the body, in conjunction with a minimal size of the latent reservoir, 136 might have significantly delayed viral load rebound in the child during the ART-free period.
Conclusions
Although residual viremia persists below the clinical limit of detection, its direct or indirect role in chronic immune activation in treated patients cannot be ruled out. If the majority of patients possess at least a portion of residual virus populations as replication-competent, the role of residual viremia in the evolution of drug resistance during suboptimal adherence to therapy and in viral load rebound after therapy interruption may turn out to be significant. On the other hand, the detection of drug-sensitive defective viruses in plasma would indicate the release of these viruses from infected cells that are stably maintained in the body during suppressive therapy. The cellular source(s) of residual viruses (being replication competent or not) may represent the “active reservoirs” for HIV-1 under suppressive therapy. Elucidating the nature and cell tropism of residual viruses and the mechanisms of their persistent production at the cellular level should remain important for future investigations, which may require the reconstruction of residual viruses from plasma vRNA of at least several successfully treated patients.
The elimination of the latent HIV-1 reservoir, which requires both effective “kick” and “kill” strategies, 138 remains a daunting challenge to the biomedical community. 139 In contrast, the naturally “active” reservoirs executing residual viremia perhaps necessitate only an effective “kill” strategy without a prior “kick” for its elimination. Therefore, residual viremia can be utilized as a unique marker in the assessment of any therapeutic candidate's ability to target and eliminate virus-expressing cells in the body. The long-lasting depletion of this marker should help reveal the success of such trials in patients on ART.
Footnotes
Acknowledgments
I am thankful to Samantha Rassler for proofreading the manuscript. This work is supported by both NIH Grant 5R03AI100722 and funding from RWMC to G.K.S.
Author Disclosure Statement
No competing financial interests exist.