
Abstract
Highly active antiretroviral therapy (HAART) is very effective in suppressing HIV-1 replication and restoring immune functions in HIV-infected individuals. However, it fails to eradicate the latent viral reservoirs and fully resolve chronic inflammation in HIV infection. The “shock-and-kill” strategy was recently proposed to induce latent HIV expression in the presence of HAART. Recent studies have shown that the protein kinase C (PKC) agonists are highly potent in inducing latent HIV expression from the viral reservoirs in vitro and ex vivo and in protecting primary CD4+ T cells from HIV infection through down-modulation of their HIV coreceptor expression. The PKC agonists are excellent candidates for advancing to clinical HIV eradication strategies. This article will present a critical review of the structure and function of known PKC agonists, their mechanisms for the reactivation of latent HIV expression, and the potential of these compounds for advancing clinical HIV eradication strategies.
Introduction
O
The “shock and kill” strategy was proposed to disrupt the viral latency to activate latent HIV expression and to target it for clearance by the anti-HIV immune response or therapeutic interventions. The histone deacetylase (HDAC) inhibitor known as suberoylanilide hydroxamic acid (SAHA) was used in clinical studies to disrupt HIV latency and activate viral expression in HIV-infected patients. 6 Another compound called JQ1 is a known agonist of the CyclinT1/CDK9 (P-TEFb) complex and was shown to be effective in disrupting HIV latency in vitro. 7 Among the various compounds tested for the disruption of HIV latency in vitro and ex vivo in the pursuit of an HIV cure, protein kinase C (PKC) agonists were found to be highly potent in inducing latent viral expression though NF-κB signaling. Therefore, PKC inhibitors are potential candidates for future clinical HIV eradication investigations.
A potential mechanism for the regulation of HIV expression by PKCs is through the phosphorylation and inactivation of IκB. Previous studies showed that mitogen [phorbol 12-myristate 13-acetate (PMA) or phytohemagglutinin (PHA)]-stimulated HIV expression in T cells involved the activation of the HIV LTR through PKC-NF-κB signaling. 8,9 Similarly, PKC-NF-κB signaling contributed to the disruption of HIV latency in the U1 monocyte cell culture model. 10 –12 Efforts to identify the specific isoforms of PKCs in the disruption of HIV latency were pursued in several cell culture models in vitro. 13 –16 The development of the Jurkat CD4+ T cell model of HIV latency facilitated the identification of a PKC agonist, prostratin, as a potent activator of HIV from latency. 17 Another PKC agonist, bryostatin-1, and its analogs are highly potent in inducing latent HIV expression and provide new opportunities for clinical investigations of HIV eradication. 18,19
Recent studies of HIV latency models in vitro and ex vivo suggest that multiple molecular mechanisms contribute to the establishment of HIV latency. Therefore, a combination of compounds targeting different mechanisms may have synergistic effects in activating latent HIV expression. Interestingly, PKC agonists by themselves are highly potent in inducing latent HIV expression. 20,21 These findings suggest that gaining a better understanding of PKC-NF-κB signaling for the disruption of HIV latency and discovering or developing new compounds for modulating this pathway should be a high priority. This review will focus on recent findings involving the role of the PKC-NF-κB signaling pathway and the potential for a new family of PKC-NF-κB agonists to disrupt HIV latency in studies focusing on an HIV cure.
Role of NF-κB in the Establishment of HIV Latency
NF-κB dimers are formed by the combination of five different monomers, p65/RelA, c-Rel, RelB, NF-κB1 (p105/50), and NF-κB2 (p100/52), which share an N-terminal Rel homology domain (RHD) responsible for dimerization, DNA binding, nuclear translocation, and interaction with IκB proteins. The p65, c-Rel, and RelB contain a transcription activation domain (TAD) and are essential for the activation of gene expression. The homodimer of p50 and p52 functions as a transcription inhibitor.
Low levels of nuclear NF-κB in resting CD4+ T cells may support the establishment of HIV latency. 22 Previous studies showed that the HIV long terminal repeat (LTR) region harbors two κB binding sites, which are highly conserved among the majority of the HIV isolates. The crystal structure of NF-κB revealed that the RelA/p50 dimer could occupy both κB sites in the HIV LTR. The two dimers clamp DNA from opposite faces of the double helix and form a topological trap of the bound DNA. 23 Upon cell activation, the p50/p65 heterodimer binds to the NF-κB sites in the HIV LTR and recruits histone acetyltransferase (HAT) to acetylate the histone tails and to open the nucleosomes to facilitate HIV transcription.
During the initial transcription, the transactivation response RNA element (TAR) is transcribed and a unique stem-like structure is formed. The viral Tat protein binds to the TAR region with the P-TEFb complex by disrupting HEXIM1. The transcriptionally active form of cyclinT1/CDK9 phosphorylates Ser-2 or Ser-5 at the C-terminal domain (CTD) of RNA polymerase II (Pol II), which increases RNA Pol II processing, resulting in high levels of HIV gene expression. In contrast, the p50/p65 heterodimer is replaced by the p50/p50 homodimer during HIV latency. This binds to the HIV LTR and recruits HDAC1 leading to the suppression of HIV expression 24 (Fig. 1).
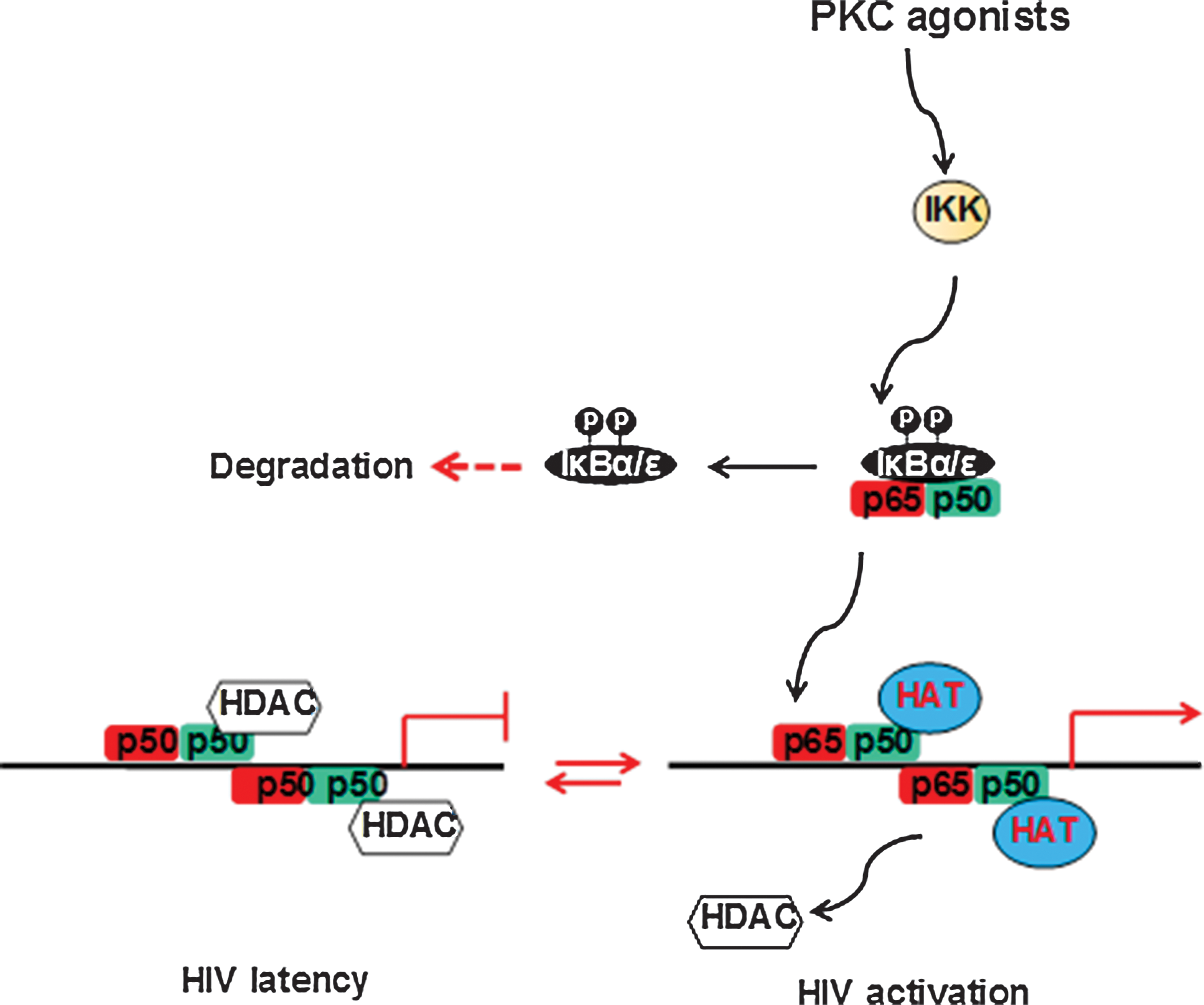
Protein kinase C-NF-κB signaling regulates HIV transcription. HAT, histone acetyltransferase; HDAC, histone deacetylase; IKK, the I kappa B kinase. Color images available online at
Recruitment of the IκBs sequesters the NF-κB dimer in the cytoplasm and prevents its translocation into the nucleus, resulting in the suppression of HIV gene expression. Following the activation of the NF-κB essential modulator protein, IκB proteins are phosphorylated by the IκB kinases, causing ubiquitination and protein degradation of IκBs, allowing NF-κB translocation into the nucleus.
There are six known IκB proteins in humans: IκBα, IκBβ, IκBɛ, IκBγ (the C-terminus of p105), IκBδ (the C-terminus of p100), and Bcl-3. The ankyrin (ANK) repeats in IκBs mediate the interaction with the NF-κB dimer (Fig. 2). Among IκBs, IκBɛ is expressed predominantly in T cells. Recently, IκBα and in particular IκBɛ were found to be critical for reactivation of HIV from latency. Therefore, targeting the inactivation of IκBɛ may present a new strategy for eliminating the latent reservoirs of HIV. 25 A search for new compounds for reactivating latent HIV reservoirs can be based on their ability to phosphorylate IκBɛ and induce IκB inactivation.
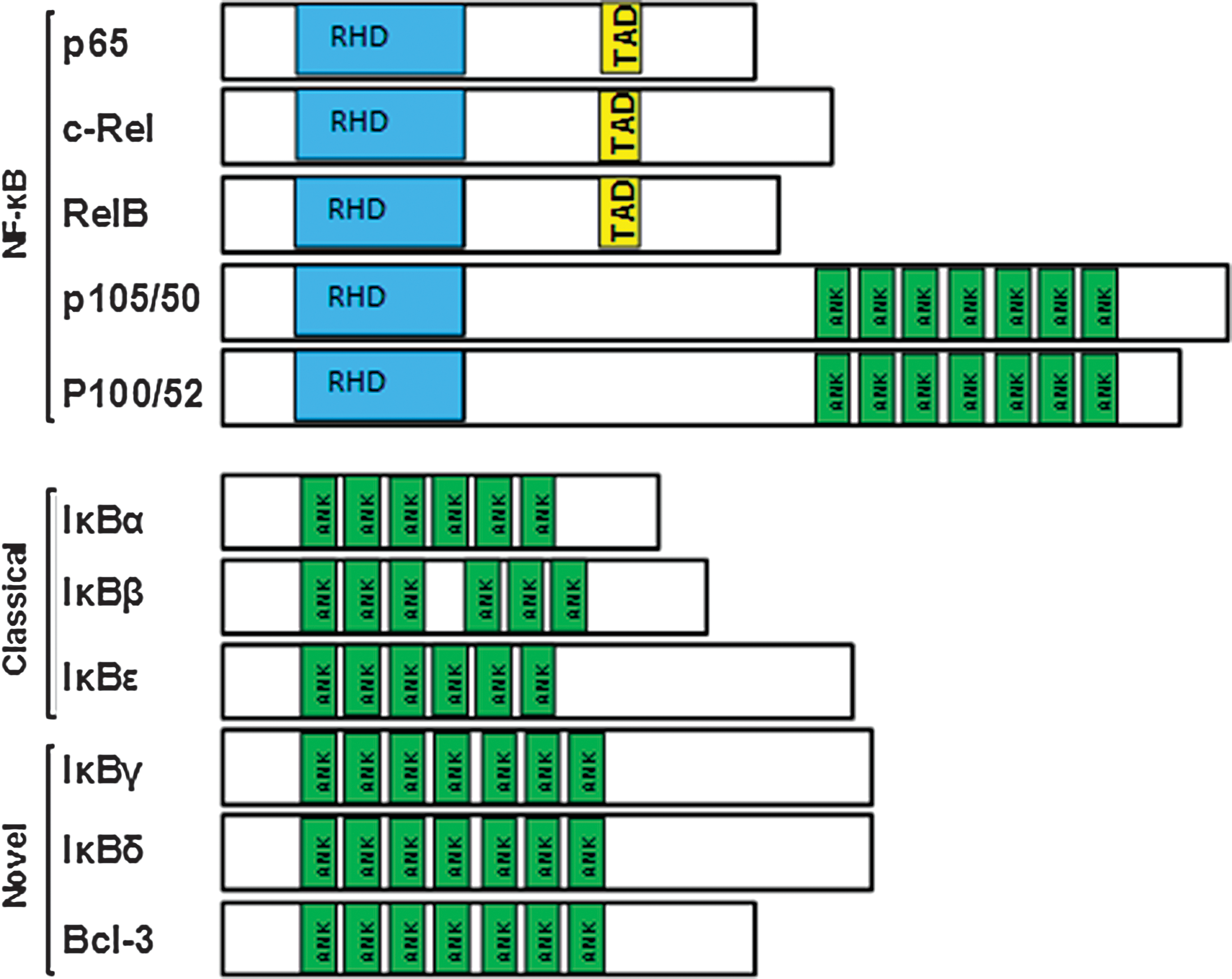
Schematic diagram of NF-κB and IκB proteins. All NF-κB proteins share a Rel homology region (RHD). The p50 and p52 isoforms also have ankyrin repeat domains (ANK), which resemble the structures found in all the IκB family. TAD, transcription activation domain. Color images available online at
The HIV subtypes harbor different numbers of NF-κB binding sites. The HIV-1 subtype E harbors one κB binding site whereas HIV-1 subtype B has two and HIV-1 subtype C has three or four κB binding sites. The number of κB binding sites in the HIV LTR promoter has been correlated with the magnitude of viral transcription in immune cells following exposure to PKC-NF-κB agonists such as tumor necrosis factor (TNF)-α, ingenol B, and PMA. Therefore, the increased number of NF-κB binding sites in the viral LTR may impact the establishment of viral predominance. 26 –31 However, it is not known whether an increased number of NF-κB binding sites in the LTR region has any impact on the kinetics of the establishment of HIV latency. High viremia with a prolonged time period has been observed in patients with primary HIV-1 subtype C infection. Therefore, it is possible that a large viral reservoir of latently HIV-infected CD4+ T cells may be established in these patients. 26 –31 Variations in the κB binding sites in subtypes of HIV may be associated with the establishment of the degree of HIV latency.
PKC Signaling for Activating the NF-κB Pathway
The PKC phosphorylation of IκB is a critical step in the activation of NF-κB. 8 Induction of HIV expression following mitogenic stimulation of immune cells occurs through NF-κB signaling. 9 The PKC-NF-κB signaling constitutes an important mechanism for the reactivation of HIV from latency. 10 –12
Three PKC families and at least 12 members were identified as important intermediate regulators for several signaling cascades and diverse cellular processes for inducing gene expression (Fig. 3). PKC proteins harbor an N-terminal regulatory domain (the C1 and C2 subregions) and a C-terminal catalytic domain (the C3 and C4 subregions). The C1 subregion mediates an interaction with phospholipids or diacylglycerol (DAG), the C3 subregion creates a cavity for ATP binding, while the C4 subregion acts mainly as a substrate-binding site. Thus, classical PKCs are regulated by lipids, DAG as well as Ca2+. Since novel PKCs lack a functional C2 subregion but contain an intact C1 subregion, they are regulated by the lipids, DAG but not Ca2+. Due to a complete lack of C2 but a partial retention of the C1 subregion, atypical PKCs are independent of DAG and Ca2+ regulation but are regulated by phospholipids.
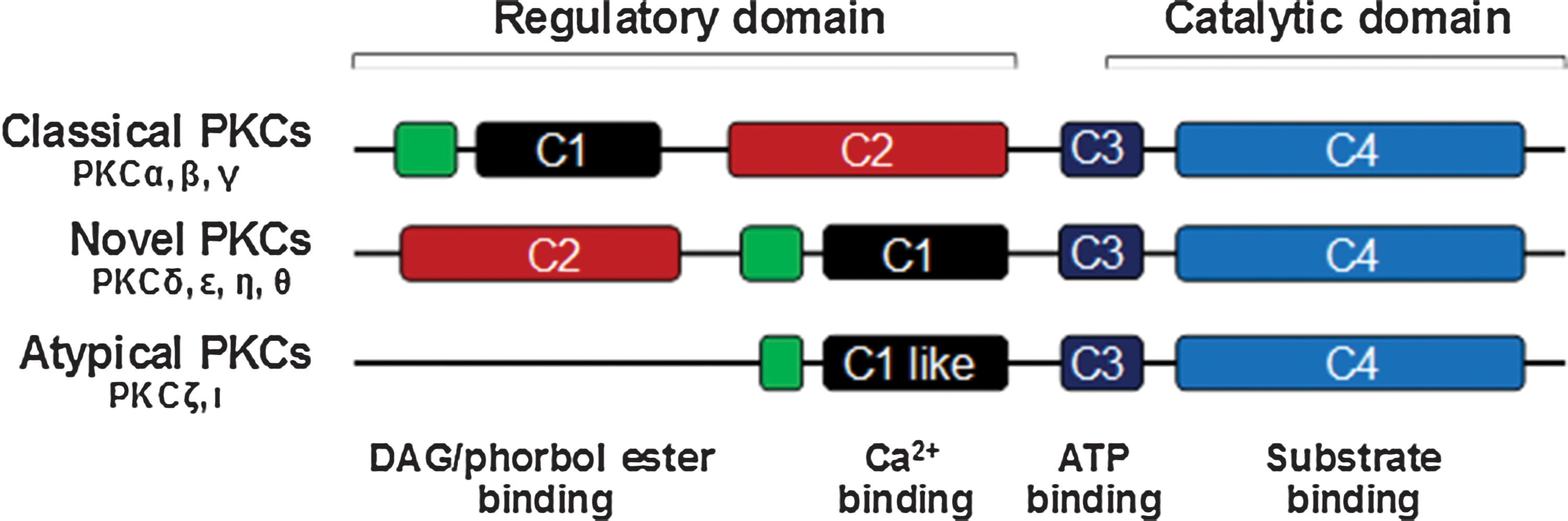
Structures of protein kinase C (PKC) families. All PKCs contain a C1 or C1-like domain that mediates their binding to diacylglycerol (DAG) or phorbol 12-myristate 13-acetate (PMA). The C2 domain in the novel PKCs does not bind to calcium. The atypical PKCs, which lack the C2 domain, are not activated by calcium, DAG, or PMA, but are by the phosphatidylserine and cis-unsaturated fatty acids. All PKC regulatory domains have a pseudosubstrate domain (shown in green) in the regulatory domain to maintain the enzymes in an inactive state. Color images available online at
There is a common region, the pseudosubstrate (PS) region, located in the regulatory domain that is shared by all PKC families. This region is critical in maintaining the PKC enzyme in an inactive state. When ligands bind to the N-terminal domain of PKCs, configuration of the protein structure releases PS from the substrate-binding site, leading to activation of the PKC enzyme and subcellular translocation of PKCs. Activation of the PKC pathway is regulated by a key family of enzymes named phospholipase C (PLC), which metabolizes phosphatidylinositol 4,5-bisphosphate [PI(4,5)P(2)] into two second messengers: inositol 1,4,5-trisphosphate [Ins(1,4,5)P(3), IP3] and DAG. The IP3 triggers the release of calcium from intracellular stores in ER into cytoplasm, where the free Ca2+ binds to the regulatory proteins and mediates multiple processes. DAG mediates the activation of PKCs in cooperation with Ca2+, leading to the phosphorylation of κB and activation of NF-κB after the hydrophobic interaction of PKC with the plasma membrane or other subcellular surfaces. 19,32
PKC Agonists for HIV Latency Research
Several natural and synthetic diterpenes were shown to activate PKC isoforms by binding to the regulatory domain, mimicking the physiologic ligand DAG and rapidly trafficking through the cell membrane to modulate PKC-NF-κB signaling to reverse HIV latency. 33 These diterpenes include phorbol esters, such as PMA, prostratin, and 12-deoxyphorbol 13-phenylacetate (DPP), and non-phorbol ester compounds including bryostatin, DAG analogs, ingenol derivatives, as well as ingol diterpenes and gnidimacrin.
Phorbol ester compounds: PMA, prostratin, and DPP
PMA is a structural mimic of DAG. However, it is oncogenic and causes transient fever and mild dyspnea in patients, even at low doses. 34 Based on the phorbol core structure, the synthesis of 12-aminoacylphorboids has been successfully achieved. These compounds reactivated more than 50% of the latent HIV through PKC signaling at a low level of 5 μM in the J-Lat cell model in vitro. However, cytotoxicity data for these compounds are not available. 35 Another novel phorbol ester, prostratin, was identified that is known to have minimal tumor-promoting activity 36 (Fig. 4).
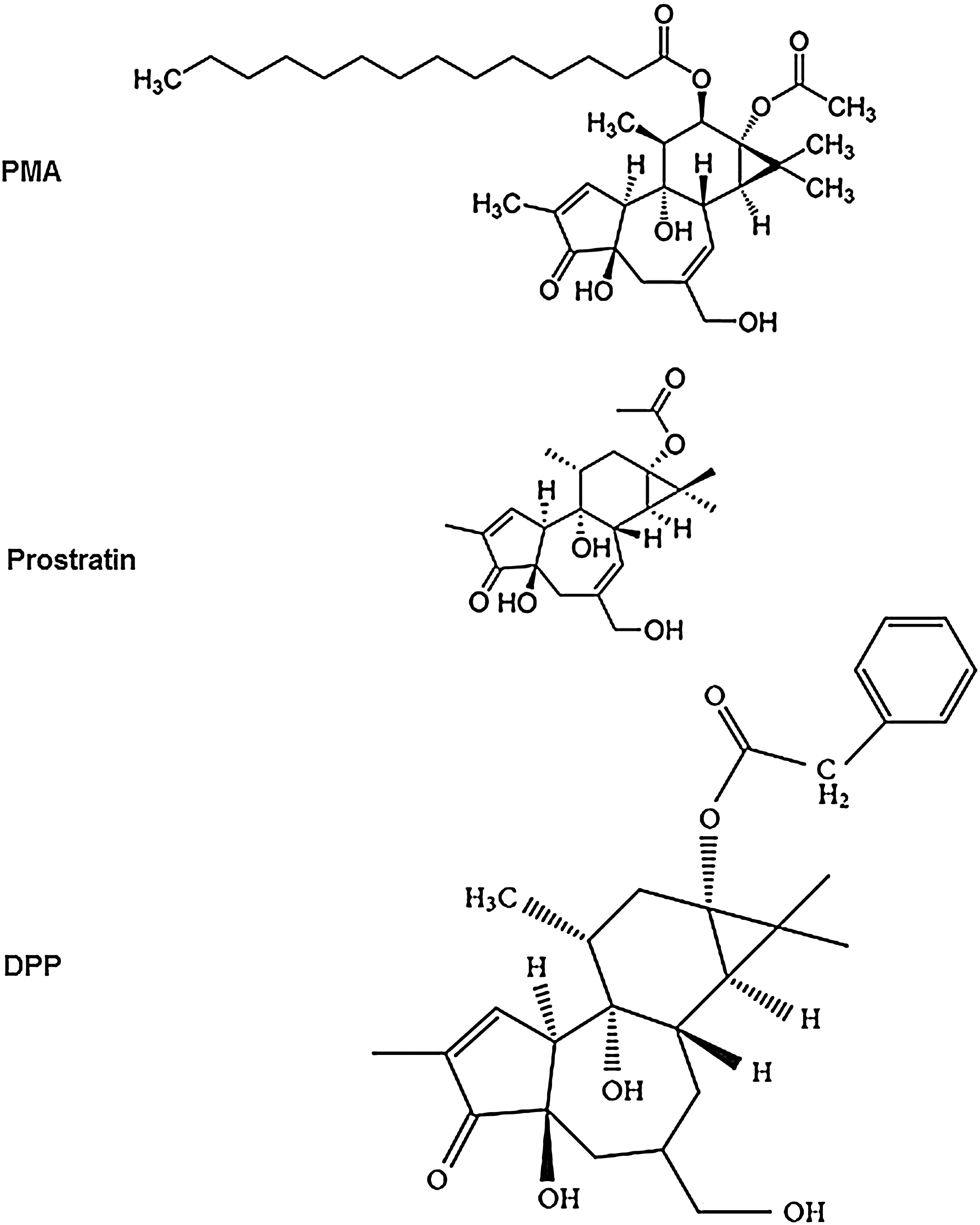
Structures of PMA, prostratin, and DPP. Prostratin and DPP share a similar core structure with PMA but show relatively less tumor-promoting activity.
Prostratin was originally isolated from a plant called Pimelea prostrata, a native of New Zealand. 37 It was later identified in the bark of the mamala tree Homalanthus nutans in Australia and was routinely used by natives in Western Samoa to treat hepatitis among local populations. 38,39 Prostratin did not stimulate proliferation of quiescent T cells at lower concentrations. However, it did increase the expression of CD69, an early T cell activation marker, while the expression of the late T cell activation marker, CD25, was not altered. 40 Although prostratin was not cytotoxic during short-term treatment, it induced substantial growth arrest and cell death in J-Lat cells when administered at a concentration of > 500 nM for more than 2 days. In zebrafish, in the 1 to 10 μM concentration range, no developmental or lethal effects were observed. 41 Therefore, only low-dose treatment of prostratin for a short period of time seemed feasible. 17
Prostratin has dual effects on CD4+ T cells that are relevant to HIV infection. It causes reactivation of HIV in CD4+ T cells harboring latent proviruses while down-regulating the expression of HIV coreceptors CCR5 and CXCR4 and preventing the spread of new HIV infection. These studies highlight the potential of prostratin as a novel therapeutic to target latent HIV reservoirs and as a candidate in clinical trials for HIV eradication.
Among the six known PKC-activating compounds, prostratin had the lowest activity for binding to PKC and translocating from the cytosol to the particulate fraction. In comparison, bryostatin 1 has displayed high binding activity to PKCδ. 42 DPP is a phorbol ester that was originally isolated from a West African plant. Like prostratin, it inhibits PMA-induced tumor promotion in CD-1 mouse skin. 43 It is highly effective in reactivating HIV provirus in cell culture models and in the HIV-infected humanized mouse model at very low nanomolar levels and about 20- to 40-fold more effective than prostratin. It also suppresses the expression of HIV coreceptors CXCR4 and CD4. 44 Clinical investigations of DPP-induced HIV reactivation are ongoing, but information regarding the toxicity and activation markers in CD4+ T cells is not yet available. 45
Bryostatin compounds
Bryostatins are a group of macrolide lactones that were originally isolated from extracts of Bugula neritina. 46 They exert antineoplastic activity against several types of cancers. Bryostatin-1 causes induction of latent HIV expression through its interaction with the PKC pathway. 42 However, bryostatin-1 seemed to cause severe myalgia and showed modest to minimal clinical benefits for patients with recurrent ovarian cancer. Many new bryostatin analogs have been designed and synthesized in order to enhance their potency 47,48 (Fig. 5).
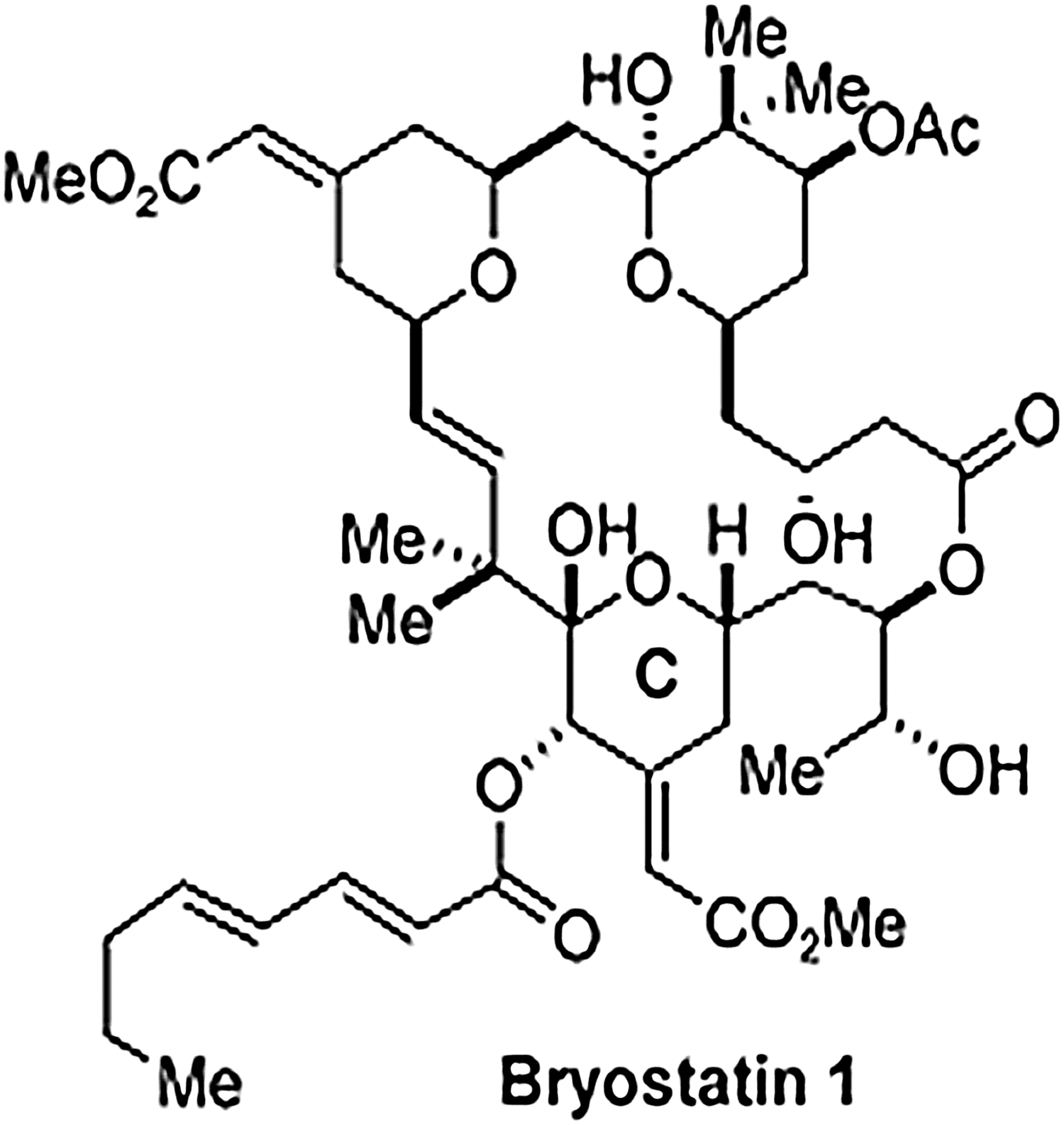
Structure of bryostatin 1. Bryostatin 1 is the lead member of the bryostatin family that displays high potency to reverse HIV latency.
Bryostatin-1 has been shown to activate latent HIV LTR in U1 cells and J1.1 cells over a range of 2.5 to 50 nmol without any significant cytotoxicity to the cells. A pretreatment of these cells with the calcium ionophore enhanced the magnitude of the bryostatin-1-induced reactivation of latent HIV by 10-fold. 11,18 Although bryostatin-1 suppressed the expression of CD4 and CXCR4 in Jurkat cells, this did not lead to the inhibition of HIV infection. Surprisingly, it did not enhance the expression of CD69 or CD25 in peripheral blood mononuclear cells (PBMCs). Bryostatin-1 is the only PKC agonist that has been evaluated for its pharmacokinetics and cytotoxicity in humans. Importantly, bryostatin-1 exhibited synergistic effects with other HDAC inhibitors, such as valproic acid (VPA). Using a newly developed ex vivo cell culture assay, Sillicano and colleagues demonstrated that bryostatin-1 alone induced a significant increase in HIV reactivation from patient-derived resting CD4+ T cells, as compared to other HIV latency disrupting agents. 21 Recently, Wender and colleagues described seven new members of bryostatin analogs that were highly potent in inducing HIV reactivation in cell culture models for HIV latency. Some of them had 1,000-fold higher potency for reactivating the virus compared to prostratin. 49 These findings provide the rationale for developing and evaluating new PKC agonists for targeting latent HIV reservoirs.
DAG lactone compounds
DAG binds to PKCs and activates classical and novel PKCs. It is metabolically unstable. DAG lactones are not immunogenic and are easy to synthesize. By modifying the branched side chains on the DAG template, increased selectivity and specificity of DAG can be engineered. Several new DAG analogs were designed and synthesized as high-affinity ligands for the C1 domain of PKCs and have shown high potency and specificity as antitumor agents by specifically activating PKCs. 50,51 Two of the analogs, LMC03 and LMC07, were shown to reactivate HIV latency not only in J-Lat cells in vitro but also in PBMCs from ART-suppressed HIV-infected patients (Fig. 6). Interestingly, like other PKC agonists, these DAG lactones also suppressed the expression of cell surface receptors, CD4 and CXCR4, but minimally up-regulated TNF-α expression. These findings suggest that DAG lactones warrant further evaluation as potential candidates for anti-HIV latency agents.

Structure of DAG lactones. The DAG lactones, LMC03 and LMC07, are synthesized using the DAG.
Ingenol compounds
Ingenol 3,5,20-triacetate (ITA) was originally isolated from a Chinese traditional medicine called Gansui, which is an extract of the dried roots of Euphorbia kansui Liou. This ingenol was found to induce HIV expression. 52,53 Some of the ingenol derivatives, including ITA and I-3-A, enhanced HIV replication at nanomolar levels in chronically HIV-infected cells, depending on or independent of the PKC/NF-κB pathway. 52,53 We recently found that ingenol-3-hexanoate (IngB), a new ingenol compound from the Brazilian plant Euphorbia tirucalli, activated latent HIV LTR in the J-Lat cells in vitro as well as in U1 cells and was more potent than SAHA, JQ1, HMBA, or prostratin in reactivation of the provirus 54 (Fig. 7).
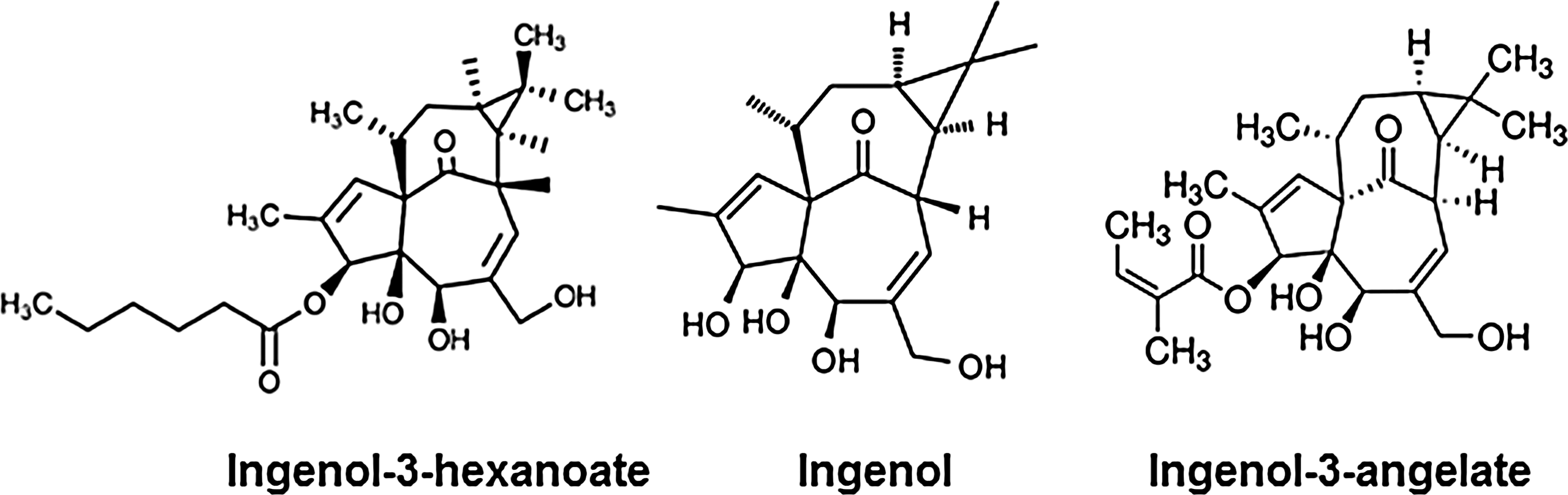
Structure of ingenols. Ingenol-3-hexanoate (IngB) and ingenol 3-angelate (I-3-A) are derived from the core ingenol by modifications at carbon 3.
IngB could reactivate latent HIV expression in J-Lat cells at a low concentration of 10–20 pM, probably through PKCδ-phospho-S664-NFκB signaling. The IngB at 3–6 nM levels had minimal cytotoxicity to CD4+ T cells with no apparent effect on cell proliferation or cell death. The same level of IngB had a minimal effect on the expression of CD38, HLA-DR, and cytokines such as interferon (IFN)-γ, interleukin (IL)-2, IL-6, and TNF-α. Importantly, even at 6 nM, IngB reactivated latent HIV expression in primary CD4+ T cells isolated from ART-suppressed HIV-positive individuals, suggesting that IngB may be a potential candidate for reversing HIV latency. 54 Similar findings about the effects of IngB on HIV replication in cell culture models of HIV latency were reported, but involved the use of higher concentrations of IngB, up to 1 μM. 55 Interestingly, IngB also inhibited HIV replication by suppressing the expression of CD4, CCR5, and CXCR4. However, IngB caused an increase in the expression of CD69, NF-κB, or P-TEFb proteins in CD4+ T cells at higher concentrations, raising some concerns about its potential side effects. 30,54,55
Gnidimacrin and other diterpenes
Gnidimacrin reactivated latent HIV LTR in U1 and ACH2 cell culture models of HIV latency at picomolar concentrations. It inhibited HIV infection of T cell lines and primary PBMCs by down-modulating the expression of HIV coreceptors CCR5 or CXCR4. Interestingly, gnidimacrin treatment caused selective killing of the chronically HIV-infected cells and warrants further investigation concerning its potency in HIV reactivation 56 (Fig. 8).

Structures of the diterpenes. The ingol diterpenes including gnidimacrin, 8-methoxyingol 7,12-diacetate 3-phenylacetate, SJ23B, and Compound 3 have a similar core structure and are potent PKC agonists.
Several studies have shown that some of the ingol diterpenes can reactivate latent HIV in cell culture models in vitro. 57 –59 The ingol diterpene 8-methoxyingol 7,12-diacetate 3-phenylacetate was isolated from Euphorbia officinarum. It caused G0/G1 cell cycle arrest in J-Lat cells without significant cytotoxicity. It induced expression in 25% of latent HIV at 10 μM concentration, showing that it was less effective than other diterpenes. 57 The second diterpene, SJ23B, was isolated from a Mediterranean plant, Euphorbia hyberna. At 0.1 μM, SJ23B induced activation of 60% of the latent HIV LTRs in J-Lat cells. Since it is not a tumor promoter, it may be a candidate for further studies. 58 The third diterpene, compound 3, was isolated from Euphorbia lactea, and it activated 80% of the latent HIV in the cells at an EC50 of 0.5 μg/ml 59 (Fig. 8). While all three diterpenes displayed a high degree of reactivation of latent HIV genomes in the cell culture models, their potency has not been evaluated in primary CD4+ T cells from ART-suppressed patients, and their cytotoxic effects on T cells have not been examined.
Conclusions and Perspectives
PKC agonists show great promise for HIV eradication using the “shock and kill” approach. They are unique because of their multiple functional characteristics. (1) They are very effective in inducing reactivation of latent HIV predominantly through PKC-NF-κB signaling. Some of them, including IngB or gnidimacrin, could activate latent HIV expression at very low picomolar concentrations. (2) While these PKC agonists cause varying degrees of T cell activation (CD25, CD38, or HLA-DR expression), they all significantly up-regulate CD69 expression, except for bryostatin-1. 18 This is not surprising since the promoter of the CD69 gene contains NF-κB binding sites. However, it is not known whether the increased expression of CD69 mirrors the magnitude of cellular toxicity. It is possible that some degree of cell activation is required for the efficacy of these compounds but may not directly translate into systematic and harmful cytokine storms. (3) Some of the PKC agonists show synergistic effects with SAHA, VPA, or JQ1 for the reactivation of HIV latency, making them strong candidates for a study of HIV cure. 54,60 (4) Most of the PKC agonists were originally isolated from traditional medicinal plants. However, currently available methodologies for the chemical synthesis of most of these compounds are efficient and economical, which makes them accessible for potential clinical use. 61 (5) An extended hydrophobic chain outward from the PKC/PMA complex likely associates with and retains the complexes at the plasma membrane, resulting in sustained PKC signaling to promote its cellular toxicity. 34 Therefore, minimizing the ancillary toxicities of PKC agonists could be achieved by altering select reactive groups.
PKC agonists have been identified as candidates for reactivating HIV from latency. Recent studies have shown the potential of PKC agonists for developing strategies for HIV cure. First, it seems that PKC agonists uniquely showed a broad capacity to reactivate HIV from latency in multiple HIV latency models. 20 Second, anti-CD3/CD38-mediated T cell activation has so far been the most effective approach for reactivating HIV-1 proviruses ex vivo (an efficacy of 7.5% producing unspliced HIV RNA by anti-CD3/CD38 compared to 0.12% using SAHA) and this occurs through PKC signaling. 62 Lastly, only the protein kinase C agonist bryostatin-1 induced significant HIV reactivation from resting CD4+ T cell reservoirs without significant T cell activation. 21 This class of compounds therefore represents an interesting possibility for the development of new anti-HIV drugs to target latent viral reservoirs in patients receiving ART. Since the establishment of HIV latency involves multiple molecular pathways, future investigations may benefit from combining the use of PKC agonists with other HIV latency-disrupting agents with different mechanisms of action.
There is an urgent need to identify HIV latency-disrupting drugs that are potent, noncytotoxic, and capable of reaching viral sanctuaries at various anatomic sites in the body. Studies of PKC agonists in HIV latency for the past 20 years or so suggest that modifying currently available PKC agonists may help achieve these goals. Therefore, it may be timely to reevaluate the structures and function of PKC agonists for research into a cure for HIV.
Footnotes
Acknowledgments
We apologize for not being able to cite all pertinent publications due to space limitation. This work was supported by NIH grants DK61297 and AI43274 and a UC Davis Research Investments in Science and Engineering (RISE) grant to S.D.
Author Disclosure Statement
No competing financial interests exist.