
Abstract
By the end of 2012, more than 6.1 million people were infected with HIV-1 in South Africa. Subtype C was responsible for the majority of these infections and more than 300 near full-length genomes (NFLGs) have been published. Currently very few non-subtype C isolates have been identified and characterized within the country, particularly full genome non-C isolates. Seven patients from the Tygerberg Virology (TV) cohort were previously identified as possible non-C subtypes and were selected for further analyses. RNA was isolated from five individuals (TV047, TV096, TV101, TV218, and TV546) and DNA from TV016 and TV1057. The NFLGs of these samples were amplified in overlapping fragments and sequenced. Online subtyping tools REGA version 3 and jpHMM were used to screen for subtypes and recombinants. Maximum likelihood (ML) phylogenetic analysis (phyML) was used to infer subtypes and SimPlot was used to confirm possible intersubtype recombinants. We identified three subtype B (TV016, TV047, and TV1057) isolates, one subtype A1 (TV096), one subtype G (TV546), one unique AD (TV101), and one unique AC (TV218) recombinant form. This is the first NFLG of subtype G that has been described in South Africa. The subtype B sequences described also increased the NFLG subtype B sequences in Africa from three to six. There is a need for more NFLG sequences, as partial HIV-1 sequences may underrepresent viral recombinant forms. It is also necessary to continue monitoring the evolution and spread of HIV-1 in South Africa, because understanding viral diversity may play an important role in HIV-1 prevention strategies.
Introduction
S
A total of 309 full or near full-length unique HIV-1 genomes from South Africa have been characterized in various studies, 296 (95.78 %) of which are subtype C isolates. 5 –10 Other South African near full-length HIV-1 genomes include two subtype A1 isolates, 8,11 two subtype B isolates, 8,11 five subtype D isolates, 12,13 and four viral recombinant forms, which included three different URF_AC recombinant forms and one complex URF. 6,8,14
We describe the near full-length genome (NFLG) sequencing and phylogenetic analysis of seven additional South African viral strains, including HIV-1 subtypes A, B, G, and two URFs.
Materials and Methods
Ethics statement
This study was approved by the Health Research Ethics Committee (HREC) of Stellenbosch University (IRB0005239) and all study participants provided written informed consent for the collection of samples and subsequent analyses.
Patients and RNA/DNA isolation
Plasma and peripheral blood mononuclear cell (PBMC) samples from the Tygerberg Virology (TV) cohort were obtained between 1998 and 2004. The TV cohort, which was previously described in Jacobs et al., 15 is a rich sample repository containing specimens from patients from wide and diverse backgrounds based on race, socioeconomic status, and sexual orientation. Viral genotyping was performed on the envelope region of 410 sequences 15 and the partial gag, pol, and env regions of a further 10 sequences. 11 A total of 35 (8.53%) non-C isolates were identified among the 410 samples from the TV cohort. Of these, seven non-C strains were selected for further characterization based on the availability and quantity of samples.
RNA was extracted from 1 ml of the plasma samples (TV047, TV096, TV101, TV218, and TV546) using the QIAamp Ultrasense Virus kit. High-molecular-weight DNA was extracted from cultured TV016 using the Qiagen DNAeasy Blood and Tissue kit and from uncultured TV1057 using the QIAmp DNA Mini kit (Qiagen, GmbH, Hilden, Germany). These two samples were genotyped from proviral DNA due to the lack of plasma in the case of TV016, the difficulty in amplifying from RNA in the case of TV1057, and the fact that these two patients were on treatment at the time of sampling.
RT-PCR and sequencing of TV047, TV096, TV101, TV218, and TV546
Between four and six overlapping fragments spanning the genome of HIV-1 were amplified using a nested long-range reverse transcriptase polymerase chain reaction (RT-PCR) method. Reverse transcription was performed with Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA) for cDNA synthesis, as described previously. 16 Primer sequences, not provided here, can be found in previous publications. 17 –19 Primers p24-7, poli8R, JH38R, and 9131R-2 were used for cDNA synthesis. For PCR amplification, primer combinations RE9737F/p24-7 (first round) and RE9745/MOp24-6 (nested PCR) were used to amplify a 1.2-kb fragment from the 5' long terminal repeat (LTR) to gag p24. Primer combinations VFgag8/LPpoli-2 and VFgag9/LPpoli-4 (nested PCR) were used to amplify a 3.8-kb fragment spanning gag p24–pol IN from four specimens and VFgag8/ppr4b and VFgag9/ppr10 from specimen TV218. Primer pairs 4274F-2 (5′ ACAGCAGTACAAATGGCAGTATTCATTC)/LP7728R and 4277F-2 (5′ GCAGTACAAATGGCAGTATTCAT)/LP7725R were utilized to amplify a 3.4-kb pol–env region from TV096, TV101, and TV546. Primer pairs ppf4/IDRps3R and ppf5B/LP7632R (5′ TATCCCATTGCAGCCAGGTCAT) were used to amplify a 3.2-kb pol IN–env IDR from TV218.
The region spanning pol–env in TV047 was amplified in two overlapping fragments: pol–env V3 (2.3 kb) using primers ppf4b/V3vh2R (5′ AAAAATTCCCCTCCACA) and ppf5b/V3vh4R (5′ GTGCRTTACAATTTCYGGGTCC) and an envV3- IDR fragment using primer pairs V3vh1F (5′ TAGGCCAGYAGTRTCAAC)/JH38R and V3vh3F (5′ GCAGTCTRGCAGAARAAGAGGTARTA)/IDRps3R. To complete coverage of the pol–env region for TV047 and TV218, an additional pol-IN product was amplified using primers and conditions as previously described. 17
A 1.6-kb env IDR-3' LTR product was generated with the primer combinations 7496F/9131R-2 and 7542F/9110R-2 from TV096 and TV218. Primer pairs JH41/9131R-2 and env-27F/9110R-2 amplified this region from TV047 and TV101, and a combination of primers env-27F/9131R-2 and 7542F/9110R-2 was used for amplification from specimen TV546.
PCR amplifications were performed with Advantage-2 polymerase Mix (Clontech, Palo Alto, CA) at cycling conditions described earlier. 16 PCR products were purified with the QIAquick PCR purification kit (Qiagen Inc.) and both strands were sequenced directly using the ABI Prism Big Dye Terminator Cycle Sequencing Reactions kit v. 1.0 (Applied Biosystems) and the ABI PRISM 3100 Genetic Analyser (Applied Biosystems). Sequence data were assembled and edited using Sequencher software Version 4.0.5 (Gene Code Corporation, Ann Arbor, MI). Positions with sequence ambiguities were assigned the appropriate IUPAC designations.
PCR and sequencing of TV016 and TV1057
Four overlapping fragments, LTR–gag (1.09 kb), gag–pol (3.89 kb), pol–env (3.94 kb), and env–LTR (1.64 kb), were amplified using GoTaq DNA polymerase (Promega, Madison, WI) as described previously. 11 Sequencing reactions were done with the ABI Prism BigDye Terminator Cycle sequencing kit v. 1.0 and run on the ABI 3130xl automated DNA sequencer (Applied Biosystems, Foster City, CA). Sequenced data were assembled into contiguous fragments and edited in Sequencher Version 4.8 (Gene Codes Corporation, Ann Arbor, MI).
Sequence quality analysis and preliminary subtyping using online tools
The HIV-1 sequence quality analysis tool was run before further analysis (
Sequences were then screened with online HIV-1 viral subtyping and recombinant detection tools: jpHMM (
SimPlot bootscan analyses
A multiple alignment was done with the HIV-1 reference subtypes (
Maximum likelihood (ML) phylogenetic tree inference
We compiled a dataset that included the HIV-1 subtype reference dataset from the Los Alamos Database, as well as randomly selected additional sequences. Multiple sequence alignments were done with Clustal W.
23
Thereafter, all of the genes of HIV-1 [with the exception of the long terminal repeats (LTR) and the nef-coding region] were concatenated in Se-Al v 2.0 (
A modeltest was performed in jModelTest 1.0 using the Akaike Information Criterion (AIC) method to estimate the best-fitting model of nucleotide substitution. Maximum likelihood tree topology was inferred in phyML v. 3.0. 24 The maximum likelihood tree topology was inferred with the GTR, an estimated Gamma shape parameter, and the subtree pruning and regrafting (SPR) method of tree rearrangement. Branch support was calculated with the implementation of bootstrap resampling totaling 100 bootstrap replicates.
Phylogenetic analyses of possible recombinants, TV101 and TV218
Based on the breakpoints identified with online tools, ML tree topologies were inferred for each of the recombinant fragments. Each fragment was aligned with the HIV-1 subtype reference alignment in Clustal W and edited in Se-Al v 2.0 as described before (
The AC recombinant NFLG sequences described previously in South Africa (AF411956, GU201611, and DQ093606) were investigated with TV218, using jpHMM to determine if the breakpoints were similar and if we had a possible new CRF.
Results
Patient demographics
All available clinical and demographic data of the seven patients are summarized in Table 1. The mean viral load was 61,608 RNA copies/ml (SD=71,246.257), while the mean CD4+ cell count was 548 cells/mm3 (SD=1,139.721). The dates of sampling collection predate the implementation of the public national HIV treatment campaign and thus only two of the patients were receiving antiretroviral therapy at the time of sampling.
M, male; MR, mixed race; F, female; Af, African; Ca, caucasian
ART, antiretroviral treatment.
Near full-length genomic sequences
Assembly of the overlapping amplification products TV47 (9,083 nucleotides), TV96 (9,058 nucleotides), TV101 (9,038 nucleotides), TV218 (9,039 nucleotides), and TV546 (9,104 nucleotides) resulted in characterization of NFLG spanning from the 5' U5 region through the 3' U3 region. Open reading frames (ORFs) were identified for gag, pol, and env structural genes and for vif, vpr, vpu, nef, tat, and rev regulatory/accessory genes. TV016 (8,039 nucleotides) and TV1057 (8,084 nucleotides) spanned from the beginning of the gag to the end of the env region and excluded the nef ORF.
Online subtyping and bootscan analyses
Preliminary subtyping was done using the online REGA version 3 and jpHMM tools, summarized in Table 2. These tools provide a quick, preliminary analysis of the sequences before detailed manual phylogenetic inference. Three isolates (TV016, TV047, and TV1057) were identified with high confidence as HIV-1 subtype B. TV096 was identified as HIV-1 subtype A1 and TV546 as subtype G. The REGA and jpHMM tools identified two possible recombinants, TV101 and TV218. SimPlot bootscan analysis identified TV101 as an A1/D recombinant and TV218 as an A1/C recombinant. TV016, TV047, and TV1057 were confirmed as HIV-1 subtype B with high similarity scores using SimPlot and TV096 and TV546 as subtype A1 and subtype G, respectively.
ML, maximum likelihood; jpHMM, jumping profile hidden Markov model.
ML phylogenetic tree inference
The ML tree is indicated in Fig. 1. Three isolates, TV016, TV047, and TV1057, clustered within HIV-1 subtype B with high bootstrap support. TV546 clustered within HIV-1 subtype G with high support, while TV218 clustered as an outlier to the subtype C cluster. The remaining two strains, TV096 and TV101, clustered within HIV-1 subtype A1 with high support. Based on the phylogenetic inference it would seem that TV218, as an outlier to the larger subtype C cluster, may represent a subtype C viral recombinant form. TV101 clustered within the A1 clade but this can possibly be explained by the small recombinant regions of subtype D that are interspersed within the larger subtype A1 sequence.
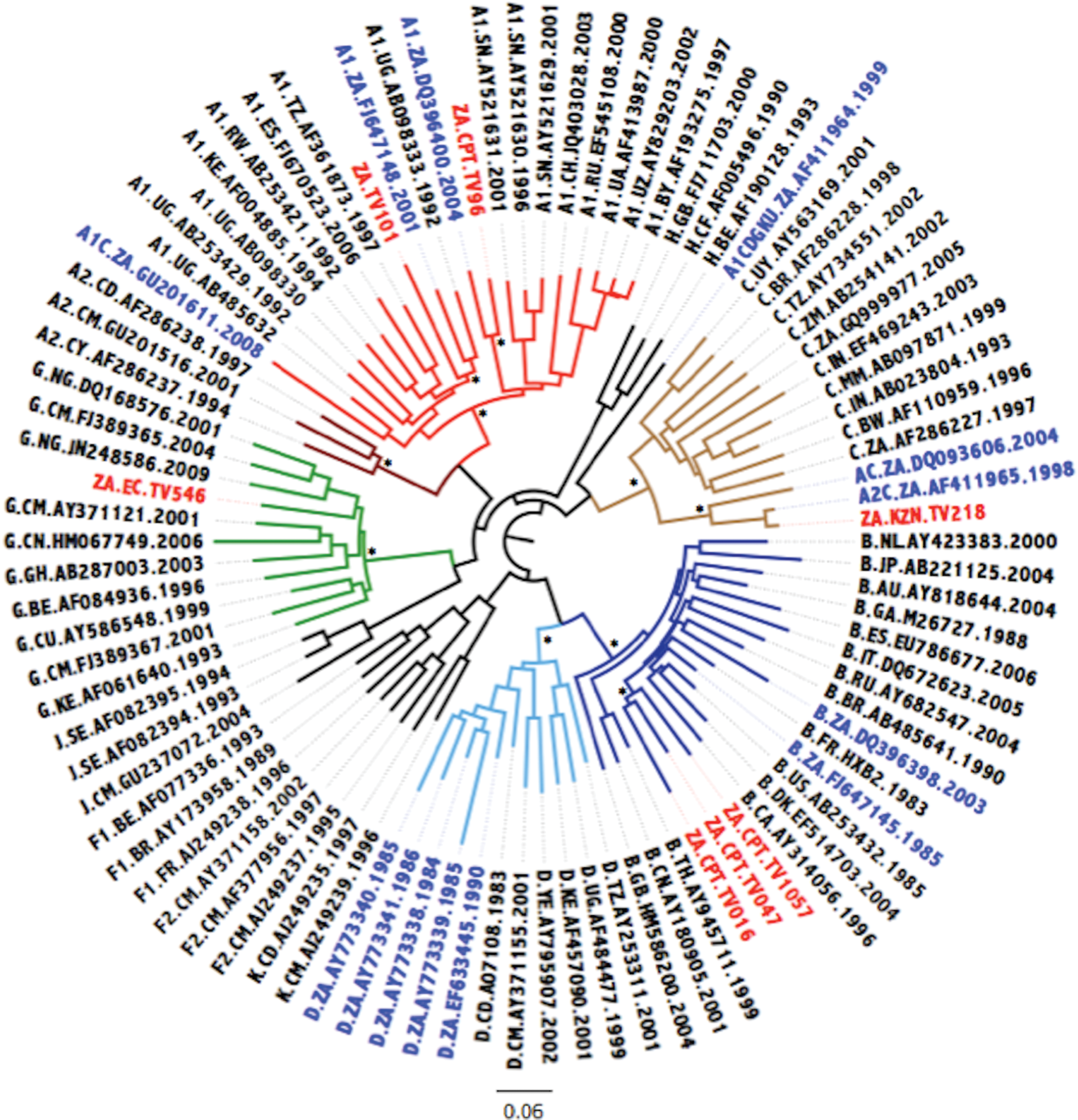
Maximum likelihood (ML) tree inference of the near full-length genome (NFLG) concatenated sequence alignment. The ML tree inferred in phylogenetic analysis (phyML) contains the seven newly sequenced Tygerberg Virology (TV) isolates, HIV-1 reference strains, and 13 previously characterized non-subtype C strains from South Africa. The evolutionary distances were computed using the GTR model of nucleic acid substitution with an estimated Gamma shape parameter. The genetic distance is displaying in the scale bar at the bottom of the figure, while the major different clades of HIV-1 Group M have been highlighted. The sequence IDs of South African strains have been marked with the 7 newly genotyped isolates marked in red and the 13 previously genotyped isolates marked in blue. Bootstrap support values for the internal branches for each major clade are shown with an asterisk and indicate support higher than 90%. Color images available online at
Phylogenetic analysis of TV101 and TV218 recombinants
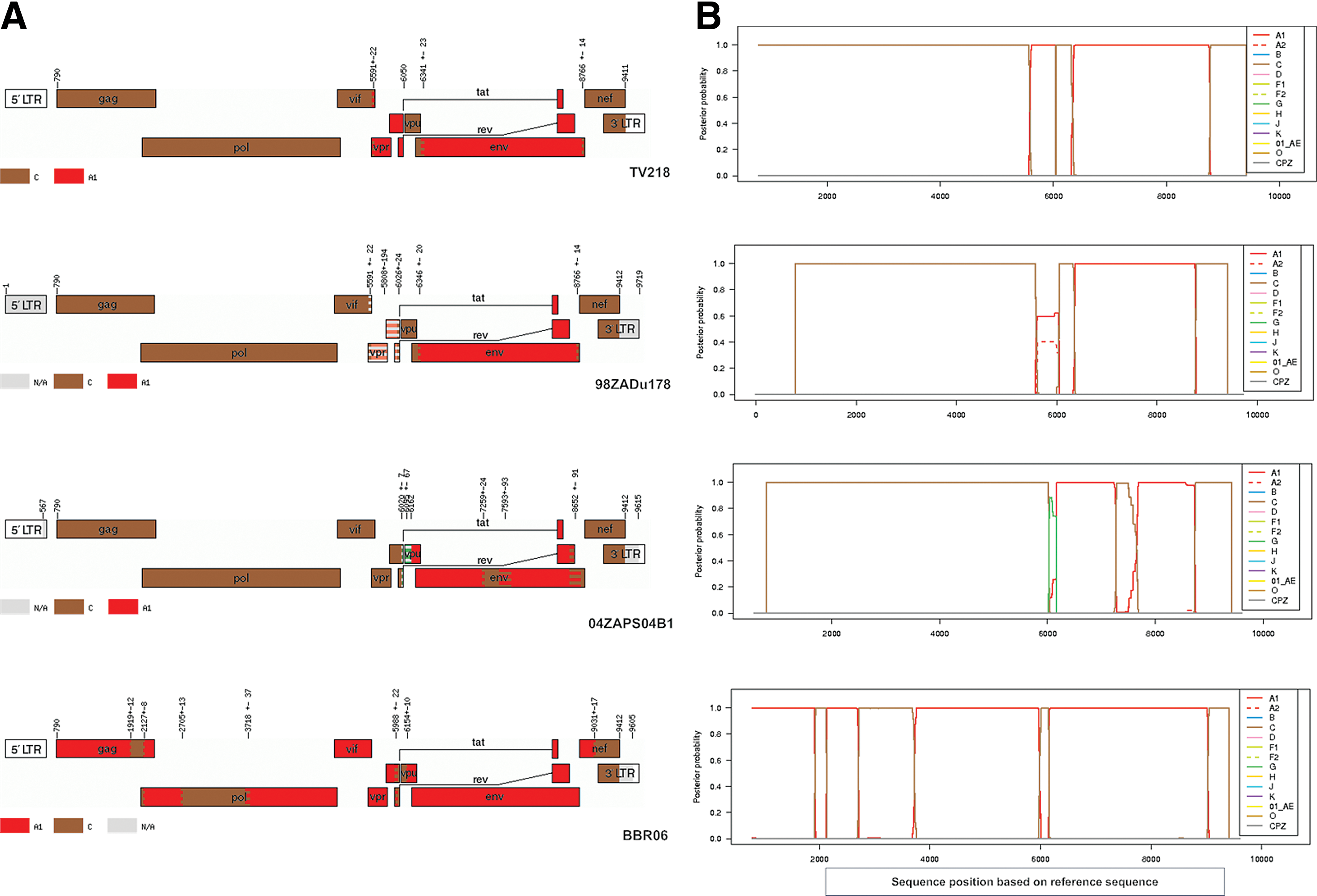
TV101 and TV218 were further investigated and phylogenies were also inferred from the recombinant breakpoints identified with online jpHMM, REGA version 3, and bootscan analyses. Representations of the genome mosaic of TV101 and TV218 are illustrated in Fig. 2 and Fig. 3, respectively. TV101 is a recombinant between HIV-1 subtype A1 and D with six breakpoints and TV218 is a recombinant between HIV-1 subtype C and A1 with four breakpoints. The breakpoints of all the other South African AC sequences are illustrated in Fig. 4.

Unique recombinant form of TV101.
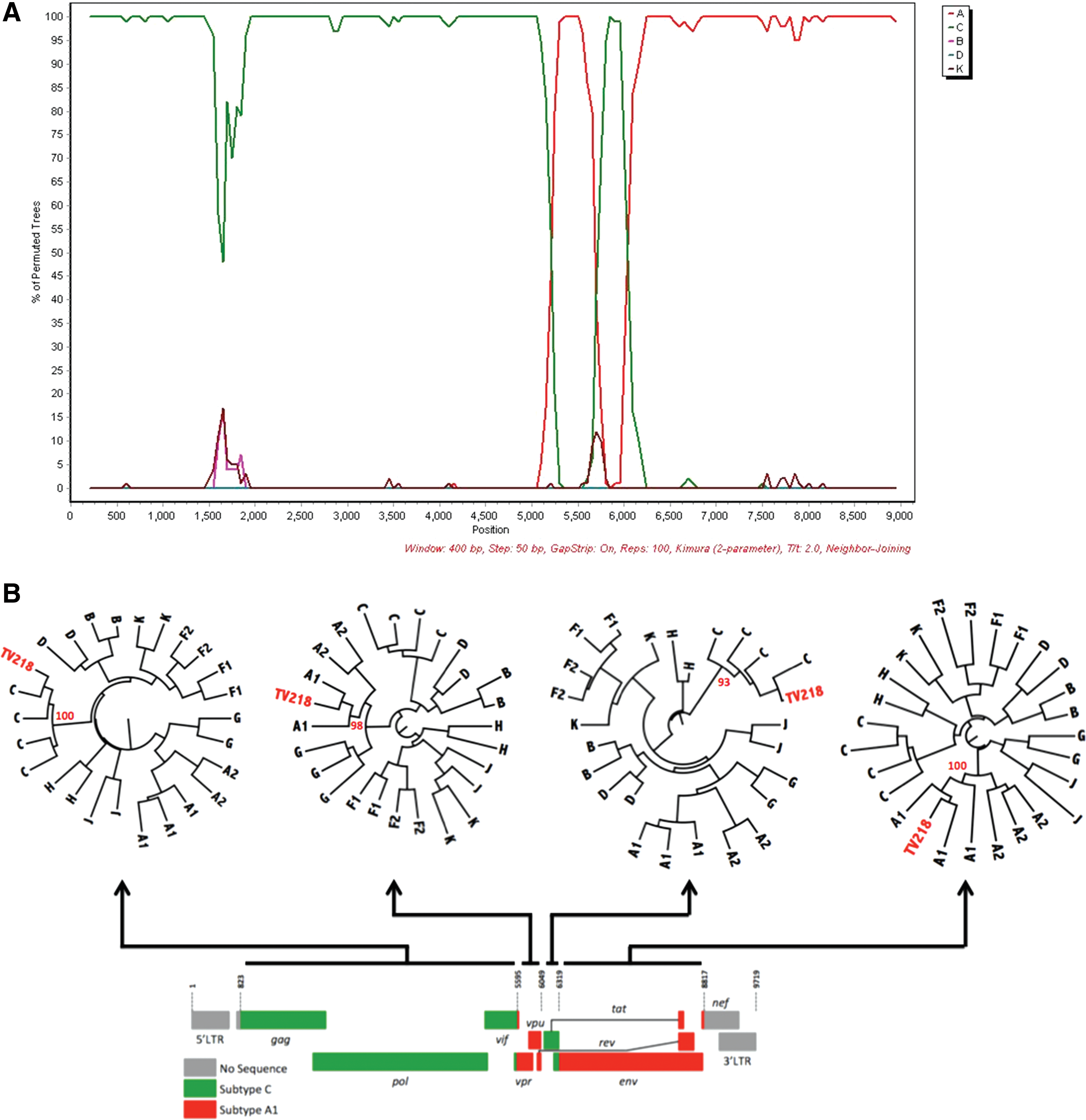
Unique recombinant form of TV218.
jpHMM analysis of South African AC recombinants.
Discussion
Limited information is available for HIV-1 non-subtype C sequences in South Africa and currently only 13 non-subtype C NFLG sequences are listed in the LANL HIV database. In this study, seven new NFLG sequences were characterized: three subtype Bs (TV016, TV047, and TV1057), one subtype A1 (TV096), one subtype G (TV546), one unique AD (TV101), and one unique AC (TV218) recombinant form. This is the first NFLG of subtype G that has been characterized from South Africa. Only three subtype B NFLGs have been described from Africa and with this article we increase this number to six.
These NFLG subtype B sequences include virus strains isolated from both heterosexual and homosexual individuals. All of these NFLGs identified in the present study were characterized in HIV-1-infected South Africans. Traditionally, subtype C has been the predominant viral form of HIV-1 in South Africa and today the subtype still accounts for the majority of infections (>95.0%). However, in recent years we have seen an increase in the number of non-C HIV-1 isolates characterized among South African individuals (E. Wilkinson and G.B. Jacobs, unpublished observations). It is of the utmost importance to continue to monitor the genetic diversity of the HIV-1 epidemic within the country, as increasing heterogeneity can potentially impact the design of an effective vaccine, viral diagnostic assays, disease progression, and treatment and may lead to the rise of more recombinant forms. There are currently more than 65 CRFs identified in the Los Alamos Database and in 2011 they were responsible for at least 20% of HIV-1 infections worldwide. 3
HIV-1 subtype B in South Africa
Previously, only two subtype B NFLGs have been characterized from South Africa 8,11 and another one from the African continent. 25 This isolate from Gabon shows no close phylogenetic relationship with any of the South African subtype B strains. Two of the subtype B strains (TV016 and TV047) were genotyped from patients who were heterosexually infected, while the other remaining patient, TV1057, became infected via homosexual contact in 1982. He is classified as a slow progressor and was receiving antiretroviral therapy when the sample was taken. Two different HIV epidemics have been described within South Africa: HIV-1 subtype B in homosexual men represented the early epidemic and accounted for the majority of HIV infections during the 1980s 26 and HIV-1 subtype C in the heterosexual population caused a later (or second) epidemic and is currently the most prevalent subtype. 27
Although TV1057 was sampled in 2001, the infection occurred in 1982, providing an opportunity to analyze a subtype B strain that originated from the time of the early epidemic in the country. This strain was most closely related to GenBank accession number EF363124 from the United States as identified through a BLAST search. TV016 (infected heterosexually in 1989) and TV047 are characteristic of an emerging subtype B epidemic occurring in the heterosexual population, indicating a crossover of the two epidemics. 15,28,29 TV016 was most closely related to GenBank accession number EF363124 and TV047 to GenBank accession number AY835795.
HIV-1 subtype G in South Africa
TV546 is the first NFLG subtype G strain that has been characterized within South Africa. The first full-length HIV-1 subtype G sequences were described in 1998 originating from the Democratic Republic of the Congo. 30 The majority of HIV-1 subtype G isolates identified originate from several West Central and East African countries. 31 –33 Subtype G is also included in nosocomial epidemic outbreaks in the former Soviet Union 34 and more recently has been linked to spread among infected drug users in the Iberian peninsula. 35,36 There has been one report of the detection of subtype G gag sequences in South Africa that was generated from samples that were obtained from migrant workers from Nigeria, Kenya, Zambia, and Angola. 37 Compared to other subtypes, subtype G occurs infrequently and TV546 is only the second report of this subtype in South Africa. The strain was detected in a female patient residing in a rural area of the Eastern Cape Province and was most closely related to isolate 944-5 from Cameroon (FJ389366) as indicated by BLAST. This patient, who became infected via heterosexual contact, had no history of travel outside of South Africa.
HIV-1 subtype A1 in South Africa
TV096 is only the third NFLG of the subtype A1 isolate characterized from South Africa. The first full-length HIV-1 subtype A1 isolate from South Africa was characterized from a female patient of African descent, 8 while the second was characterized from an African male. 11 TV096 was sampled from a female patient of African descent in the late stages of HIV infection, as characterized by World Health Organization (WHO) criteria. Blast and phylogenetic inference indicated that TV096 was more closely related to African HIV-1 subtype A1 isolates from Senegal and Uganda. Phylogenetics of the previously characterized A1 sequences from South Africa also showed a close genetic relationship with other HIV-1 subtype A1 isolates from the East African region.
Unique recombinant forms in South Africa
Two unique recombinant forms were also identified in this study. TV101 is the second URF_AD described from South Africa and is most closely related to AF457082 from Kenya. The first URF_AD recombinant was characterized from a South African individual who became infected via heterosexual contact in Kenya. 11 These two URF_AD sequences share no recombinant breakpoints. HIV-1 subtypes D and subtype A1 have been detected in South Africa in the past. 8,11,12
In addition to the URF_AD, TV218, a URF_AC was also characterized in the present study. This is the fourth URF composed of subtypes A and C that has been identified in South Africa. Two A1/C recombinants, isolate 04ZAPS204B1 (GenBank accession DQ093606) 8 and isolate BBCR06 (GenBank accession GU201611), 14 and one A2/C recombinant, 98ZADu178 (GenBank accession AF411965), 6 have already been described. TV218, 98ZADu178, and 04ZAPS204B1 were sampled in Durban, KwaZulu-Natal, while the other A1/C isolate was sampled in the far northern part of South Africa. 14
TV218 revealed a close genetic similarity to a previously described A2C subtype (AF411965, isolate 98ZADu178) from South Africa. 6 The 98ZADu178 sequence was derived from cultured cells from an asymptomatic sex worker in Durban (sample date 1998), while TV218 was directly amplified and sequenced from plasma obtained during 2000 from a 25-year-old female in Durban. These two A1C sequences are 97% similar and share similar breakpoints. The vpr, tat, and rev region of TV218 is subtype A1 with a high probability, whereas this region in 98ZADu178 is A1/A2 with a low probability.
Conclusions
Phylogenetic inference of seven newly sequenced HIV-1 strains identified subtypes A1, B, and G as well as URF_AC and URF_AD. There is a need for more NFLG sequences because partial HIV-1 sequences may underrepresent viral recombinant forms. It is necessary to continue monitoring the evolution and spread of HIV-1 in South Africa and worldwide. Understanding HIV-1 diversity in South Africa will play an important role in HIV-1 prevention strategies.
Sequence Data
The sequences analyzed during the study have been deposited in GenBank and are available under the following accession numbers: KJ948656 to KJ948662.
Footnotes
Acknowledgments
This study was funded by the Poliomyelitis Research Foundation (PRF), the National Research Foundation (NRF), and the Medical Research Council (MRC) of South Africa. This research project was funded by the South African Medical Research Council (MRC) with funds from the National Treasury under its Economic Competitiveness and Support Package. This research and the publication thereof are the result of funding provided by the Medical Research Council of South Africa in terms of the MRC's Flagships Awards Project MRC-RFA-UFSP-01-2013/ UKZNHIVEPI.
Author Disclosure Statement
No competing financial interests exist.