
Abstract
The rare hepatitis B virus (HBV) genotype G (HBV/G) coinfects HIV-1-positive individuals along with HBV/A and generates recombinants. However, the circulation of HBV A/G recombinants remains poorly understood. This molecular epidemiologic study examined HBV A/G recombinants in Japanese HIV-1-positive men who have sex with men (MSM). Initially, blood specimens submitted for confirmatory tests of HIV infection in Osaka and Tokyo, Japan, from 2006 to 2013 were examined for HIV-1, and HIV-1-positive specimens were screened for HBV. Among 817 specimens from HIV-1-positive individuals, HBsAg was detected in 59 specimens; of these, HBV/Ae (alternatively A2), a subgenotype of HBV/A prevalent in Europe and North America, was identified in 70.2%, HBV/C in 17.5%, and HBV/G in 10.5%, and HBV/E in 1.8% according to the core gene sequence. The full-length genome analysis of HBV was performed on HBV/G-positive specimens because some HBV A/G recombinants were historically overlooked by genotyping based on a partial genome analysis. It revealed that five of the specimens contained novel Ae/G recombinants, the core gene of which had a high sequence similarity to HBV/G. Detailed analyses showed that novel recombinants were coinfected with HBV/Ae in a recombinant-dominant fashion. No major drug-resistant mutations were found in the newly identified HBV Ae/G recombinants. Some of the individuals asymptomatically coinfected with HIV/HBV suffered mild liver injury. This study demonstrated that novel Ae/G HBV recombinants were identified in Japanese HIV-1-positive MSM. The pathogenicity of novel HBV Ae/G recombinants should be examined in a future longitudinal study. Surveillance of such viruses in HIV-1-positive individuals should be emphasized.
Introduction
H
HBV is classified into 10 genetic groups, termed HBV/A to J, based on an intergroup divergence of >8%. 3 –9 Each genotype was divided into subgenotypes. The genotypes of HBV show a distinct geographic distribution 10,11 and are associated with different clinical outcomes, responses to treatment with interferon or nucleotide analogues, and rates of fulminant hepatitis. 12 –21
Historically, 96.9% of HBV isolates from Japanese individuals with chronic hepatitis B belong to genotypes B or C 14 ; however, since 2000, HBV/A has spread in urban areas through homosexual intercourse. 18,21 –23 The positivity rate for HBV infection in Japanese HIV-1-infected individuals is 8.9%, and half of HBV-infected individuals harbor the genotype A virus. 24 A previous report showed that up to 90% of HBV isolates obtained from HIV-1-infected individuals were genotype A. 25 Our previous surveillance study focusing on high-risk populations that attended primary sexually transmitted infection clinics in Osaka revealed that 60% of the HBV genotypes infecting HIV-1-positive individuals were Ae (alternatively, A2), a distinct genetic cluster within HBV genotype A that was distributed in Europe and North America. 26,27 In that study, we noticed that three HIV-1-positive individuals were coinfected with HBV/G, a rarely identified genotype of HBV.
Genotype G is an unusual variant of HBV and little is known about its epidemiology, natural history, and clinical data. 28 In 2000, a unique HBV isolate harboring a 36-base pair insertion in the core region was identified in France; this was the first isolate of genotype G. 6 The S gene of HBV/G is highly homologous (94.6–97.5%) with that of HBV/A at the nucleotide (nt) level. 29 Subsequently, HBV/G was identified in the United States, 30,31 Mexico, 32 Germany, 33 Canada, 28 and Brazil. 34 A previous study estimated the prevalence of HBV/G in these areas to be 1–5%. HBV/G infection occurred predominantly in males (92%) and was primarily associated with homosexuals. 28 Indeed, HBV/G-positive individuals were coinfected with HBV/A or a recombinant genotype A/G virus, 28 which is consistent with other reports, including our own. 24,26
Recently, a number of reports documented homologous recombination between different HBV genotypes. 1 A concern is how frequently such recombinations occur, and whether the pathogenicity of recombinant HBVs differs from that of other HBV genotypes. Because HBV/G coinfects with HBV/A, genetic recombination between these genotypes is possible. Indeed, five A/G recombinants have been identified to date: two in the United States (AB056516 30,35 and JQ707426 36 ), two in Canada (EU83389 and EU83390 28 ), and one in Brazil (EF464099 34 ).
Until now, genotyping of HBV has been based on only a part of the viral genome. Importantly, analysis of the complete genome sequences of HBV isolates revealed that a viral clone previously classified as genotype G was actually an A/G recombinant. 30,35 In our previous study, our classification of the HBV genotype was not based on the full viral genome sequence, raising concerns that some A/G recombinant forms might have been overlooked. Thus, we revisited the clinical specimens to ascertain whether any A/G recombinant forms of HBV were detectable upon full genome analysis.
Materials and Methods
Specimens and serological diagnosis
Specimens found to be HIV-1 positive in the confirmatory tests were subjected to screening for the HBs antigen (Ag). A total of 817 specimens, 813 from Osaka and four from Tokyo, collected either at health care centers or medical institutions between 2006 and 2013, were analyzed.
An HIV serodiagnostic screen was performed using the Genedia HIV-1/2 mix particle agglutination (PA) anti-HIV assay method (Fujirebio, Inc., Tokyo, Japan). Confirmatory tests for HIV-1 infection were performed using LAV BLOT 1 (Fujirebio). Serological tests for HBV were performed using the Espline HBsAg kit and the SERODIA-anti-HBs PA or Espline HBsAb-N kits, which detect anti-HBs antibodies (Ab) (Fujirebio). Serological tests for HBV were also performed using the Mycell anti-rHBc diagnostic reagent, which detects anti-HBcAb (Institute of Immunology Co. Ltd., Tokyo, Japan). Serodiagnosis of Treponema pallidum (TP) infection was performed using the Serodia TP-PA test (Fujirebio).
Biochemistry
Plasma levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), lactate dehydrogenase (LDH), and gamma-glutamyl transpeptidase (γ-GTP) were measured. All specimens were diluted 2- to 20-fold in phosphate-buffered saline (PBS) prior to measurement.
Viral load tests
The HIV-1 viral load was measured using AmpliPrep/Cobas TaqMan HIV-1 v2.0 (Roche Diagnostics, Manheim, Germany) or by real-time polymerase chain reaction (PCR) according to the method described by Kondo et al. 37 The HBV viral load was measured using the COBAS AmpliPrep/Cobas TaqMan HBV Test v2.0 (Roche).
Analysis of the HIV-1 genome
The HIV-1 genome was analyzed as described previously.
38
Briefly, the env C2-V3 and pol regions of the HIV-1 genome were analyzed and the HIV-1 subtype was determined by phylogenetic analysis using GENETYX-MAC Ver. 14 (GENETYX, Tokyo, Japan). HIV drug resistance was determined using the HIVdb Program in the HIV drug resistance database (
Analysis of the HBV genome
DNA was extracted from HBsAg-positive sera using a QIAamp UltraSens Virus Kit (QIAGEN, Dusseldorf, Germany), and the HBV DNA was PCR amplified. The primers were WA-L, WA-R, HBVnext, and AS2330 (WA-L, WA-R, and AS2330 have been described previously) (Table 1).
39,40
PCR products were electrophoresed in 1% agarose gels. The DNA bands were extracted and sequenced using the BigDye Terminator kit (Applied Biosystems, Foster, CA) and the following primers: WA-L, WA-R, HBVnext, AS2330, FA1-L, FA2-L, FA3-L, FA4-L, FA1-R, FA2-R, FA3-R, FA4-R, and 1868 (Table 1).
39,40
Nucleic acid sequencing was performed in a ABI3130 automated sequencer (Applied Biosystems). Nucleic acid sequence analysis was performed using BLAST, CLUSTAL W (DDBJ: DNA Data Bank of Japan), GENETYX-MAC Ver. 14 (GENETYX, Tokyo, Japan), Molecular Evolutionary Genetics Analysis (MEGA) 5.2.2 and the Oxford HBV Automated Subtyping Tool (
According to the AB014370 coordinate.
HBV genotype-specific PCR and cloning of PCR products
Nested PCR was performed to clarify coinfection by distinct HBV genotypes. Briefly, the initial PCR was performed using the HBVALL9/HBVALL12 primer set to generate products of 1,615 and 1,651 bp, representing genotypes Ae and G, respectively (Table 1). The second PCR was performed using the HBVAEF5/HBVAER7 or HBVGF6/HBVGR8 primer sets, yielding products of 387 bp or 423 bp corresponding to HBV genotypes Ae or G, respectively (Table 1). The PCR products were confirmed by sequencing. Additionally, nested PCR primers were designed to amplify genomic DNA specific for HBV/Ae, HBV/G, and Ae/G recombinants (see Table 1).
The initial PCR was performed using the HBVALL9/HBVALL10 primer set, which amplifies all HBV genotypes. The second PCR was performed using the HBVAEF1/HBVAER3, HBVGF2/HBVGR4, or HBVAEF1/HBVGR4 primer sets, which yielded amplicons only when the template contained the Ae, G, or Ae/G genotypes, respectively. To determine the ratio of coinfecting HBV genotypes, the PCR products primed by HBVALL9/HBVALL10 were cloned into the pTAC-1 vector and sequenced (BioDynamics Laboratory, Inc., Tokyo, Japan). The amplicon was scored HBV genotype G-derived when it contained the 36-bp signature sequence unique to HBV genotype G. To analyze the genome sequence of HBV/Ae that was coinfected with the HBV Ae/G recombinant, the PCR products primed by AEF14/AER3 were sequenced.
Ethical considerations
This study was approved by the Ethical Review Board of Osaka Prefectural Institute of Public Health (approval numbers 0703-06, 0810-4, and 0810-5-2).
Results
Initial screening of HIV-1-positive individuals for HBV genotype G
We examined 817 blood samples collected from HIV-1-positive individuals from 2006 to 2013. The primary screen employing an immunochromatographic test for HBs Ag identified 59 positive specimens. HBV DNA was successfully amplified from 57 of these specimens by PCR targeting the core region of the viral genome. Sequencing analysis revealed that the most common HBV genotype was Ae (40/57 specimens, 70.2%), followed by C (10/57 specimens, 17.5%), G (6/57 specimens, 10.5%), and E (1/57 specimens, 1.8%). We focused our attention on six specimens that were positive for HBV genotype G, since this genotype is rarely detected worldwide. PCR successfully amplified the complete HBV genome from five of the specimens. These were named Os/JP/2008, Tk/JP/2009, Os/JP/2010, Os/JP/2011, and Os/JP/2013. All five individuals infected with HBV/G were men who have sex with men (MSM).
Identification of novel Ae/G recombinants of HBV
Phylogenetic analysis of the full length viral genome sequences revealed that Os/JP/2011 and Os/JP/2013 formed a distinct cluster under the genotype A group, and Os/JP/2008, Tk/JP/2009, and Os/JP/2010 formed a distinct cluster under the genotype G group (Fig. 1). However, we noticed that previously isolated Ae/G recombinants positioned in the neighboring branches. Genetic structure analysis and gene-by-gene analysis revealed that all the viruses initially judged as genotype G according to C gene sequences were A/G recombinants (Fig. 2 and Supplementary Figs S1–S4; Supplementary Data are available online at

Phylogenetic analysis of novel hepatitis B virus (HBV) A/G recombinants and comparison with representative strains from each HBV genogroup. Genetic distances were estimated using the full length genome sequences of HBV strains and the Kimura two-parameter matrix method. Phylogenetic trees were drawn using the neighbor-joining method. Accession numbers and clone names are shown on each branch. Bootstrap values are shown along with each main branch. A scale bar indicates the degree of nucleotide divergence. The novel Ae/G recombinants isolated in this study are boxed. Viral strains marked with an asterisk are reference strains representing genotypes Ae and G (Fig. 2).
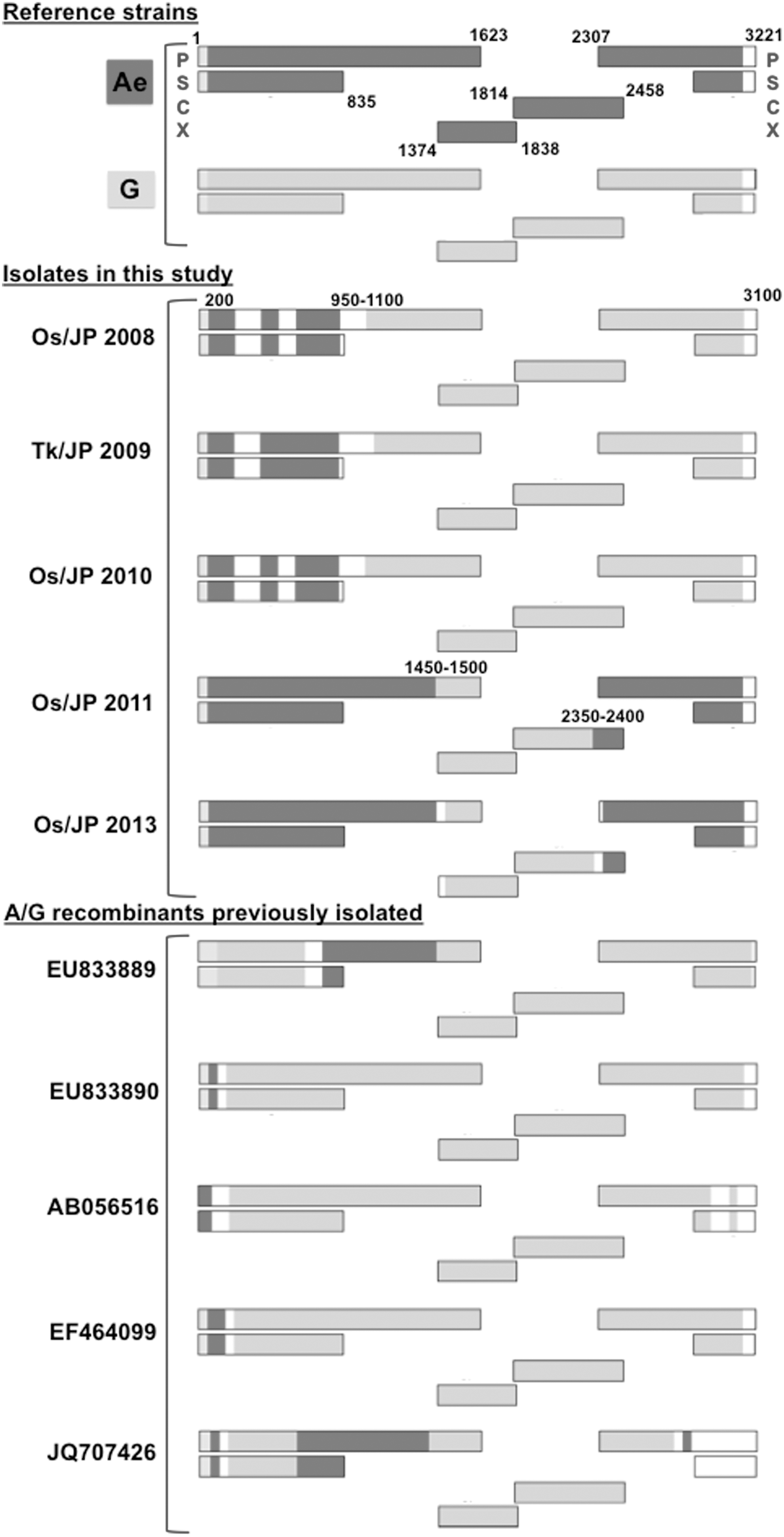
Genetic structures of the novel HBV Ae/G recombinants isolated in this study. The genomes of HBV/Ae and HBV/G are shown in dark gray and gray, respectively. Bootscan analysis was performed with a window size of 400 and a step size of 50. The representative strains of Ae and G are AB697507 and AB375170, respectively (asterisks in Fig. 1). The approximate locations of the recombination breakpoints are shown. All five previously isolated A/G recombinants are shown for reference. P, polymerase gene; S, surface gene; C, core gene; X, the X gene.
Phylogenetic analyses of P and S (nt 2848–3215, 1–835) genes revealed that Os/JP/2008, Tk/JP/2009, and Os/JP/2010 were positioned close to HBV/G strains, but Os/JP/2011 and Os/JP/2013 were clustered close to HBV/A strains (Supplementary Data S1 and S2). In contrast, all five were clustered with HBV/A or HBV/G strains when phylogenetic analyses were performed on S (nt 1–835) or C (nt 1814–2491) and X (nt 1374–1835) genes (Supplementary Data S3). Os/JP/2008, Tk/JP/2009, and Os/JP/2010 were positioned close to previously identified Ae/G recombinants and showed similar patterns of recombination, i.e., the recombination points occurred at nt 950–1100 and nt 3100–3200, spanning the 3′ end and the middle section of the S gene, respectively. The crossover patterns of Os/JP/2011 and Os/JP/2013 were distinct from those of Os/JP/2008, Tk/JP/2009, and Os/JP/2010 in that the recombination points lie at nt 1450–1500 and nt 2350–2400 at the 5' end of the X gene and at the 3′ end of the C gene, respectively (Fig. 2).
Among the five novel recombinants, Os/JP/2008, Tk/JP/2009, and Os/JP/2010 shared >99.9% sequence similarities at the nucleotide level, and showed similar recombination patterns. In contrast, Os/JP/2011 and Os/JP/2013 shared a high sequence similarity (>99.1%), although their patterns of recombination were different from those of Os/JP/2008, Tk/JP/2009, and Os/JP/2010. The env nucleotide sequence similarity of HIV that was coinfected with Os/JP/2008 was 93.8% of that with Os/JP/2010. However, the env gene of HIV-1 that was coinfected with Tk/JP/2009 had 81.4% and 82.2% sequence similarities with those with Os/JP/2008 and Os/JP/2010, respectively. Furthermore, the sequence similarity of HIV-1 env between Os/JP/2011 and Os/JP/2013 was 83.8%.
Genetic traits of the Ae/G recombinants
The genetic features of HBV/G are as follows: the presence of stop codons at codons 2 (TAA instead of CAA) and 28 (TAG instead of TGG) in the precore region; a 36-bp insertion in the core gene; mutations in the core promoter (A1762T, G1764A); and a 3-bp deletion in the pre-S1 region. All the five Ae/G recombinants identified herein shared the above genetic features. In contrast, the characteristic feature of HBV/Ae, namely a 6-bp insertion at the 3′ end of the core gene, was present only in Os/JP/2011 and Os/JP/2013.
The “Ae genotype regions” (nt 173–926) within Os/JP/2008, Tk/JP/2009, and Os/JP/2010 were genetically similar to that of an isolate identified in the United States in 1998 [accession number JQ707544]. The same genetic region within Os/JP/2011 and Os/JP/2013 was similar to that of an HBV/Ae isolate identified in Japan in 1993 [accession number KC836877]. Interestingly, the nucleotide sequences of HBV/Ae strains that coinfected with Tk/JP/2009, Os/JP/2010, Os/JP/2011, and Os/JP/2013 were highly similar to KC836877 (99.9–100%). We were not able to carry out the analysis on Os/JP/2008 due to a lack of sample.
Coinfection status of the Ae/G recombinant with HBV/Ae and /G
A previous study has shown that HBV A/G recombinants often coinfect with HBV/A and HBV/G. 28 Thus, we next examined whether HBV/A and HBV/G were present in specimens that were positive for Ae/G recombinants. To do this, we developed a PCR system that distinguished genotypes Ae, G, and Ae/G. As expected, all five specimens gave positive results in the Ae/G-specific PCR. Similarly, all five were positive in the Ae-specific PCR. However, the G-specific PCR failed to amplify any of the specimens, although the limit of detection remained to be determined. On the other hand, another PCR designed to amplify the “G genotype region” of Ae/G recombinants was positive for all five specimens that targeted the 36-bp insertion of the core gene. The specificity of PCR was verified by sequencing the amplicons. This suggests that HBV recombinant genotype Ae/G coexisted with Ae, whereas the level of genotype G was below the limit of detection.
To estimate the Ae to Ae/G recombinant ratio, we cloned and sequenced the PCR products amplified using primer set HBVALL9/HBVALL10, which amplifies both genotypes. The number of independent clones tested for Os/JP/2008, Tk/JP/2009, Os/JP/2010, Os/JP/2011, and Os/JP/2013 was 13, 2, 21, 27, and 11, respectively. All the cloned amplicons were derived from Ae/G recombinants, and no amplicons encoding HBV/Ae were found. These data suggest that Ae/G recombinants are circulating dominantly in the infected individuals.
Pathophysiology of Ae/G recombinant HBV infection
We next examined the pathophysiological characteristics of individuals harboring Ae/G recombinants (Table 2). As described above, all five Ae/G recombinant-positive individuals were Japanese MSM who were coinfected with HIV-1. They were aged 24–41 years at the time of serodiagnosis and showed no apparent clinical signs, suggesting that all were in the asymptomatic phase of HIV/HBV coinfection.
Anti-Treponema pallidum antibody.
IU, international unit. Normal range is shown.
M, man; 3TC, lamivudine; ETV, entecavir; TFV, tenofovir; RT, reverse transcriptase; MSM, men who have sex with men; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase; LDH, lactate dehydrogenase; γ-GTP, gamma-glutamyl transferase; ND, no date.
Virological analysis revealed that all were infected with HIV-1 subtype B, a common subtype in Europe, the United States, and Japan. The HIV-1 viral load in the plasma ranged from 3.3×103 to 7.7×104 per ml, which is not very high; however, the HIV-1 infection was not controlled as none of the subjects was taking antiretroviral drugs. The following mutations were identified in the HIV genome: T69N in reverse transcriptase and L10V in the viral protease. These mutations represent common polymorphisms within the HIV-1 genome, although they confer only weak resistance to reverse transcriptase or protease inhibitors.
All specimens were positive for HBsAg and HBcAb, but negative for HBsAb, suggesting that all subjects had an active HBV infection. The HBV viral load in the plasma ranged from 3.59×107 to 2.27×1010 copies per ml, indicating high levels of HBV replication. No mutations conferring resistance to the nucleoside analogues lamivudine, entecavir, or tenofovir were detected, consistent with the fact that all subjects were treatment naive.
Plasma samples were tested for parameters related to liver function, including AST, ALT, ALP, LDH, and γ-GTP. Most were within the normal range, suggesting that none of the five subjects suffered severe liver dysfunction at the time of diagnosis, although mildly increased AST or AST, ALP, and γ-GTP levels were observed in the subjects infected with Os/JP/2013 or Tk/JP/2009.
Discussion
HIV-1 was shown to spread in a specific community with high-risk behaviors, including MSM. Such individuals have high probabilities of being coinfected with HIV-1 and HBV based on the similarity of their infection route. In fact, the seroprevalence of HBV among HIV-positive individuals was 63.2%. 26 The coinfection risk of multiple HBV genotypes is also high. In Canada, 67% of HBV/G-positive individuals were male homosexuals and were coinfected with an HBV/A or A/G recombinant. 28 In Japan, the coinfection of HBV/G and /A has also been reported. 24 In individuals coinfected with distinct HBV genotypes, a recombinant HBV may be generated. HBV recombinants originating from HBV genotypes A and D or B and C have been already reported. An intergenotype HBV recombinant made from genotypes A, C, and G is now categorized as a distinct genotype I. The crossover patterns of recombinant viruses vary 1 ; however, some regions of the HBV genome appear to favor intergenotype or intragenotype recombination.
In line with previous reports, the crossover points of the five novel Ae/G recombinants described herein were positioned at or close to genetic regions in which recombinations occur frequently. 42 According to the recombination patterns, it seems likely that Os/JP/2008, Tk/JP/2009, and Os/JP/2010 originated from the same ancestor and the ancestor of Os/JP/2011 and Os/JP/2013 was different from that of the other three. Considering the high-risk behavior and the molecular epidemiological data, it was suggested that HIV-1 and HBV were not always cotransmitted in the MSM populations. We described five novel HBV Ae/G recombinants in Japanese MSM coinfected with HIV-1, and the recombination patterns of these recombinants were totally distinct from five HBV Ae/G recombinants identified previously. These data suggest that novel recombinants should have been generated independently in distinct MSM communities. It was noted that HBV genotyping using only a part of the viral genome may be misleading. 43
HBV/G, which often infects homosexual individuals, is also present in individuals harboring HBV/A. 6,30,34,44,45 This is partly due to the poor replicative capacity of HBV/G. The presence of HBV/A significantly increases the rate of HBV/G replication in hepatocytes, although infection with HBV/G alone is possible. 46 –49 Genetic rearrangements between HBV/A and HBV/G can occur during persistent coinfection. Therefore, we examined how novel Ae/G recombinants were generated and transmitted in the Japanese MSM population. To do this, we focused our analysis on genetic regions derived from HBV/Ae, but not from HBV/G, since the genetic diversity of the HBV/G genogroup is poor. Molecular epidemiological data suggested that Os/JP/2008, Tk/JP/2009, and Os/JP/2010 evolved from an ancestral Ae/G recombinant in North America that was then introduced into the Japanese population. In contrast, the ancestor of Os/JP/2011 and Os/JP/2013 viruses may be generated independently in the domestic MSM population, which was then spread along with the parental domestic strain of HBV/Ae.
The PCR experiments showed that HBV Ae/G recombinants were present dominantly over the HBV/Ae, and the levels of HBV/G were under the limit of detection in all five cases, although these data had some limitations involving the number of examined amplicons as well as intrinsic problems in PCR. However, the dominance of the Ae/G recombinant was probably due to its replicative capacity and/or selective advantage in vivo. It was likely that host immunity provided a primary selection pressure. A similar phenomenon was reported previously wherein a selection pressure may be partly attributed to antiviral agents. 36 The finding that HBV/Ae was present, albeit at a much lower frequency, suggests that Ae/G recombinants still require HBV/Ae for efficient replication, as does HBV/G.
Novel HBV recombinants were found in the specimens collected for the anonymous HIV-1 testing. Thus, we have limited clinical information about the examinees. Knowing these limitations, biochemical findings indicated that coinfection of the HBV Ae/G recombinant and HIV-1 did not always result in a rapid progress of disease in a short period of time. Some HBV genotypes show a severe clinical course. 50 Here, we were unable to determine whether these novel Ae/G HBV recombinants are more pathogenic than other HBV genotypes. This is primarily because the specimens were collected during the asymptomatic phase of HIV/HBV infection and the clinical course of the infected individuals was not monitored after virus detection. This will be addressed in a future study.
A previous report from Brazil identified HBV/G harboring mutations conferring lamivudine resistance (L180M and M204V). It is notable that two of the three reported cases were HIV positive. 34 It is a concern that the potential spread of novel Ae/G recombinants among Japanese HIV-positive MSM may result in them acquiring drug-resistant mutations that limit treatment options; however, we found no major drug-resistant mutations in any of the five novel Ae/G recombinants identified herein. Taken together, the results of the present study suggest that the molecular epidemiology of HBV should be studied in more detail, especially in HIV-1-positive individuals.
Sequence Data
GenBank accession numbers for the full genome sequences obtained in this study are as follows: AB933279–AB933283. GenBank accession numbers for the Ae sequences obtained in this study are LC029424–LC029427.
Footnotes
Acknowledgments
This study was supported by JSPS KAKENHI Grant 24590840 and Grants-in-Aid from the Ministry of Health, Labour and Welfare of Japan, grant number 25190701.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.