
Abstract
HIV-1 Tat is a regulatory protein that plays a pivotal role in viral transcription and replication. Our study aims to investigate the genetic variation of Tat exon 1 in all subtypes of HIV-1: A, B, C, D, F, G, H, J, and K. We performed phylogenetic, mutation, and selection pressure analyses on a total of 1,179 sequences of different subtypes of HIV-1 Tat obtained from the Los Alamos National Laboratory (LANL). The mean nucleotide divergences (%) among the analyzed sequences of subtypes A, B, C, D, F, G, H, J, and K were 88, 89, 90, 88, 86, 89, 88, 97, and 97, respectively. We revealed that subtype B evolved relatively faster than other subtypes. The second and fifth domains were found comparatively more variable among all subtypes. Site-by-site tests of positive selection revealed that several positions in all subtypes were under significant positive selection. Positively selected sites were found in the acidic domain at positions 3, 4, and 19, in the cysteine-rich domains at positions 24, 29, 32, and 36, in the core domain at position 40, and in the basic domain for the rest of the positions for all subtypes. Positions 58 and 68 in the basic domain were positively selected in subtypes A, B, C and B, C, F, respectively. We also observed high variability within positively selected sites in amino acid positions. Our study findings on HIV-1 Tat genetic variability may contribute to a better understanding of HIV-1 evolution as well as to the development of effective Tat-targeted therapeutics and vaccines.
Introduction
M
HIV-1 has three structural and six regulatory proteins. Many of those proteins are the targets for developing antivirals and vaccines. HIV-1 Tat, one of the regulatory proteins, is considered a target for developing vaccines 4,5 and therapeutics 6,7 as Tat plays a pivotal role in viral transcription. In a clinical trial, an HIV-1 Tat B cell epitope vaccine showed promising results in reducing HIV-1 viral loads; notably, the vaccine was designed with an aim to inhibit circulating Tat protein. 8 However, Tat binds to the transactivation-responsive region (TAR) RNA for transactivation, therefore antivirals have been designed to inhibit Tat-TAR RNA interaction. 9 Tat has two exons: exon 1 and exon 2. Exon 1 consists of five of the six domains of HIV-1 Tat; those five domains are important for the functionality of the protein.
As reported previously, genetic variation of Tat causes functional differences in its cytotoxic T cell-mediated immune response. 10 Genetic monitoring of Tat is thus important in the development of Tat-targeted vaccines and therapeutics. However, the intersubtype variation of Tat may also have an impact on its functional variation. It is therefore imperative to understand the intersubtype variation of Tat in the currently circulating strains. It has already been reported that there is high genetic variation of HIV-1 Tat in major subtypes, such as B and C. 11 However, genetic variation of HIV-1 Tat in minor subtypes remains elusive. It is therefore worthwhile to understand the extent of the genetic variation of Tat not only in major subtypes but also in minor subtypes in order to develop effective antivirals and vaccines. Therefore, our current study aims to determine the genetic variation of Tat exon 1 in all subtypes of HIV-1 M: A, B, C, D, F, G, H, J, and K. We performed phylogenetic, mutation, and selection pressure analyses among those subtypes to gather more comprehensive information regarding the genetic variability of HIV-1 Tat.
Materials and Methods
Sequence data
HIV-1 Tat sequences were obtained from the Los Alamos National Laboratory (LANL) HIV sequence database
12
using the following options: Alignment type: Web (all complete sequences); Year: up to 2013; Organism: HIV-1; Region: TAT (exon 1); Subtype: All subtypes belonged to M (except A–K recombinant); DNA/Protein: DNA; Format: FASTA. According to the LANL website, the curated alignments were published in the year after the sequences became available; therefore the “up to 2013” alignment contains all sequences published before 2014. On July 3, 2014, 2,156 sequences were initially downloaded using the “one sequence per patient” option. We excluded problematic sequences as defined by LANL's prebuilt search criteria. Sequences likely to have been contaminated with laboratory strains were also excluded through this process. As recommended by LANL, we further checked to ensure that none of the sequences contained ambiguous nucleotides, hypermutations, minor insertions, minor deletions, premature termination codons, or different sequences from the same strains (verified by the strain name). Through this sorting process, we made a study data set of a total of 1,179 sequences for subtypes A to K, which contained 71 amino acids encoded by 213 nucleotides (nt) from positions 5831 to 6045 nt in exon 1 of the HIV-1HXB2 genome (GenBank accession no. K03455). The sequence data were obtained together with information about the host, subtype, isolation year, and isolated country (Supplementary Table S1; Supplementary Data are available online at
A multiple-sequence alignment program, Clustal W, was used to align the nucleotide sequences without any gap. Consensus sequences were generated by aligning circulating primary isolates and selecting the most common nucleotide at each position. In this study, the three most common subtypes were B, C, and A, shown here as 50.7% (n=598), 28.5% (n=336), and 10.9% (n=128) of the sequences obtained, respectively. The following amounts of the total number of sequences were for other subtypes: D (4.4%, n=52), G (2.5%, n=30), F (2.3%, n=27), H (0.3%, n=4), J (0.2%, n=2), and K (0.2%, n=2). It is worthwhile to mention that this distribution of HIV-1 subtypes reflects the publicly available and published sequence data, not the global prevalence of subtypes that constitute the HIV pandemic.
Phylogenetic analysis
The phylogenetic tree was constructed using the neighbor-joining method. Evolutionary distances were computed following the Tamura–Nei
13
method and are in units representing the number of base substitutions per site. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories). The tree was drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted using Molecular Evolutionary Genetic Analysis (MEGA) version 6.
14
The reliability was estimated from 1,000 bootstrap replicates. The simian immunodeficiency virus (SIV) sequence, CPZ.US.85.US_Marilyn.AF103, was used as the outgroup to root the tree. FigTree v.1.3.1 (available at
Selection pressure analysis
After selecting the best-fit model from the possible time-reversible models, we performed the analyses using Datamonkey, an online selection pressure analysis tool. 16 –18 We calculated the overall strength of selection by comparing the relative ratio (ω) of nonsynonymous (dN) to synonymous (dS) substitutions averaged over the entire alignment (24). Three likelihood methods, fixed effects likelihood (FEL), interior branches fixed effects likelihood (iFEL), and single-likelihood ancestor counting (SLAC), were used in order to identify the existence of positive selection pressure at the whole gene level as well as at individual codon sites. We specified significance levels, p=0.05 and Bayes factor=50, per gene per site dN/dS according to at least one of the above-mentioned assay methods (SLAC, FEL, and iFEL).
Results
Phylogenetic analysis of HIV-1 Tat exon-1
To reveal the sequence identity, we performed distant analysis using the maximum composite likelihood method. Sequence identities and mean nucleotide divergence for all subtypes of HIV-1 Tat are summarized in Table 1. In phylogenetic analysis (Fig. 1), the evolutionary distances of different subtypes of Tat exon 1, estimated by the neighbor-joining (NJ) method, showed that subtype C was more closely related to the CPZ.US.85.US_Marilyn.AF103 strain than other subtypes. From that analysis, it was apparent that subtype B evolved relatively faster than the other subtypes. We also revealed that subtypes B and D were more closely related to each other than to other subtypes; notably, subtype D is considered an early clade of the subtype B African variant. The genetic distance between the rest of the subtypes and subtype C was small, especially between subtype A and subtype C. There was no evident monophyletic distribution by year of isolation or by country in any subtype, which may indicate that the infections from subtypes were transmitted in a diverse and heterogeneous manner.

Phylogenetic tree for the HIV-1 Tat exon 1. The neighbor-joining phylogenetic tree of Tat exon 1 sequences in different subtypes of HIV-1 is shown. The evolutionary distances were computed using the Tamura–Nei method with a GTR+Γ5 nucleotide substitution model in the 1,000 bootstrap replicates, using MEGA 6; the tree was visualized by FigTree v1.4.0. Branch colors were added to indicate different subtypes. SIV sequence, CPZ.US.85.US_Marilyn.AF103 was used as the outgroup to root the tree (as shown by the light green line). Color images available online at
Mutation analysis
We found the following percentages of conserved positions in different subtypes: subtype A, 42%; subtype B, 27%; subtype C, 23%; subtype D, 42%; subtype F, 55%; subtype G, 59%; subtype H, 69%; subtype J, 87%; and subtype K, 89%. Overall, subtypes B and C exhibited more genetic variation compared to other subtypes (Fig. 2 and Supplementary Table S2).
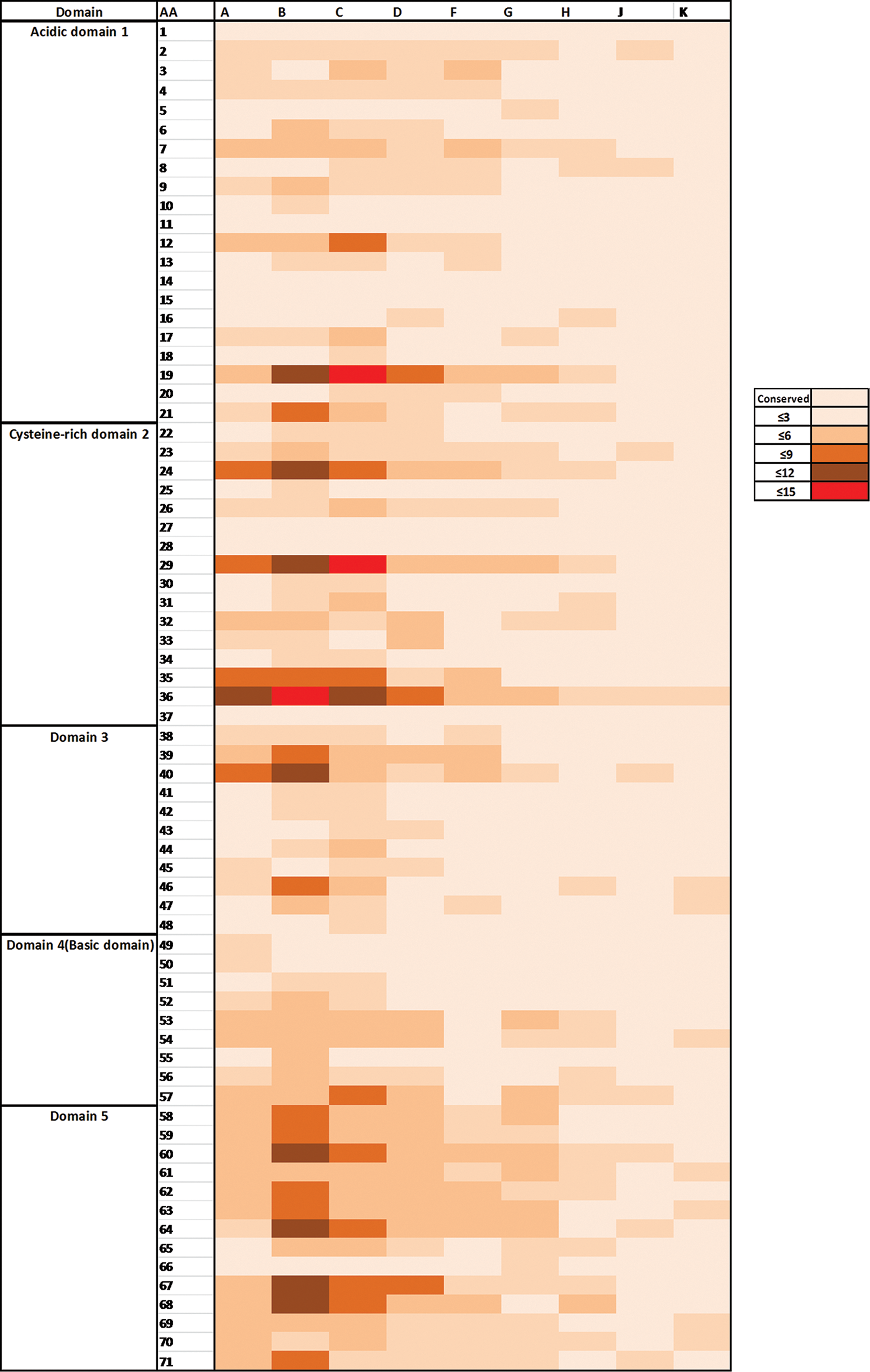
Amino acid variation in the Tat exon 1 in different subtypes. Rows indicate the amino acid position according to HIV-1HXB2 numbering and columns indicate different subtypes. The intensity of colors reflects the number of amino acid variations in each position. Color images available online at
In the first domain (residues 1–21), we found that most of the positions were conserved in all subtypes. Subtypes B, C, A, and F contained more substituted amino acids, especially at positions 7, 12, 19, and 21. At positions 7 and 12, the predominant amino acid was asparagine in all subtypes except subtype B where arginine and lysine were predominant, respectively. Notably, positions 7 and 12 are well-recognized antigen-binding sites. 10 At position 19, all subtypes except B and C mostly contained lysine, whereas there are 13 and 10 different types of amino acid substitutions present in subtypes B and C, respectively. Moreover, at position 21, the predominant amino acids were alanine and proline in all subtypes except subtype F, which contained proline, and subtypes J and K, which contained alanine only.
The second domain (residues 22–37) was the most variable region for all subtypes. At position 24, commonly found amino acids were lysine and asparagine in subtypes A, B, C, D, F, and G, asparagine in subtype H, and, interestingly, glutamine in subtypes J and K. However, more variations were observed in that position in subtypes A, B, and C. At position 29, amino acid changes were commonly observed, and most of the subtypes contained mainly lysine except subtype C, which contained histidine, and subtypes J and K, which contained arginine. At position 32, phenylalanine was present in all subtypes except H, J, and K, where it was tyrosine. At position 36, a large number of variations were observed, particularly in subtypes B, C, and A, in which valine was commonly found; in other subtypes, it was leucine.
In the third domain (residues 38 to 48) and the fourth domain (residues 49 to 57), most of the positions were relatively conserved except 39 and 40. Among the studied subtypes, variation of amino acids was mostly observed in subtype B.
The fifth domain (residues 58 to 72) was found to be comparatively more variable in all subtypes. All positions except position 65 (mainly in subtypes B, C, D, and H) and 66 (in all subtypes) were relatively conserved. Positions 58 to 60, 62 to 64, 67, and 68 were found to be the most variable in subtype B.
Selection pressure analysis
The relative ratios (ω) of nonsynonymous (dN) to synonymous (dS) substitutions for the nucleotide sequence of Tat of all subtypes except subtypes H, J, and K are shown in Fig. 3. For subtypes H, J, and K, selection pressure analysis could not be performed because of an insufficient number of eligible sequences. Interestingly, subtype F had the highest ratio value, followed by subtype B, then subtype G. In contrast, site-by-site tests of positive selection revealed that several positions in all subtypes were under significant positive selection based on the FEL, iFEL, and SLAC methods (Table 2). Positively selected codons with statistical significance calculated using the aforementioned methods are shown in Supplementary Table S3.

Selection profiles of the Tat exon 1 gene in different subtypes. The dN/dS (ω) values were calculated using the single-likelihood ancestor counting (SLAC) method. The x-axis shows different subtypes and the y-axis shows dN/dS (ω) values.
In subtypes H, J, and K, selection pressure analysis could not be performed because of an insufficient number of an eligible sequences.
According to HIV-1HXB2 numbering.
Single-likelihood ancestor counting (SLAC) method.
Fixed effects likelihood (FEL) method.
Interior branches likelihood (iFEL) method.
Positively selected sites were found in the acidic region at positions 3, 4, 19, in the cysteine-rich region at positions 24, 29, 32, and 36, in the core domain at position 40, and in the basic domain for the rest of the positions for all subtypes. Positions 58 and 68 in the basic domain were selected in subtypes A, B, C and B, C, F by all three methods, respectively. Position 70 was also commonly selected only in subtypes B and C. In subtype D, positions 19 and 62 were positively selected; these two positions were also selected in subtype A. There was no positively selected position found with the SLAC method in subtype F. However, positions 3 and 68 were selected using the FEL and iFEL methods. For subtype G, position 29 was positively selected by all three methods. Relative amino acid frequencies at positively selected codon positions determined using the SLAC, FEL, and iFEL methods in different subtypes are shown in Fig. 4.

Amino acid variation in positively selected sites in different subtypes of HIV-1 Tat exon 1. The frequency of amino acid variations in each positively selected codon position determined by single-likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), and interior branches fixed effects likelihood (iFEL) in different subtypes (subtypes A–G) is shown.
Discussion
In our study, the different subtype distributions of Tat exon 1 were broadly similar (phylogenetically) to estimated distributions of other genes such as Gag, Pol, and Env; notably, those sequences were obtained using the Los Alamos sequence compendium published in 2012. 19 We observed a 15% to 22% variation among the Tat sequences of nine HIV-1 subtypes. However, as reported previously, the genetic variation in Env amino acid sequences was about 25% to 35% among those nine subtypes, 20 whereas in Gag and Pol, the genetic variations were approximately 14% 21 and 22% to 23%, 22,23 respectively.
It is apparent in all trees that the branch lengths of the trees are not influenced by sequence length but rather by the density of nucleotide variation in each gene irrespective of the phylogenetic method used. In other words, the faster a gene evolves, the longer the branch lengths will be versus a more slowly evolving gene over the same time interval. The evolving pace of a gene is governed by several factors, most importantly by mutation rate and selection pressure. The variability in subtype distributions of HIV-1 Tat may increase over time as HIV-1 transmission in different regions of the world continues because of diverse risk behaviors and the high mobility of the infected people. 24 Notably, subtype-specific differences in transmissibility and pathogenesis may not always lead to significant changes in subtype distribution in different populations; rather, those differences might be ecologic. 25 Therefore, the association between specific subtypes and a unique mode of transmission has yet to be established.
The genetic complexity of the global HIV-1 epidemic is increasing due to the continuous molecular evolution of existing subtypes. 26 We observed high variability within positively selected sites possibly due to the fact that those sites are under diversifying selection. Notably, positive selection signifies genetic evolution and changes in antigenic sites for the binding of a virus to a host cell and also for exerting the host immune response against the infection. 27 In our analysis, the majority of selected amino acid residues were found in domains 2 and 5 of HIV-1. Domain 2 is known as the highly conserved cysteine-rich domain containing cysteine at seven positions. As revealed by previous studies, any amino acid change in this domain affects the phenotypic characteristics of Tat since this region is responsible for making disulfide bonds 28,29 and regulating CCR5 expression in monocytes. 30 Domain 5 plays an important role in cell surface binding and internalization, and any changes in this domain also influence the functionality of Tat. 31,32 Relatively few positively selected variants were detected at the N terminus of Tat exon 1 between codons 1 to 21, and the TAR binding site, a functionally important region. In addition to being conserved and negatively selected, those regions contain several experimentally defined cytotoxic T-lymphocyte (CTL) epitopes. 10 Taken together, the amino acid changes of those two domains may have functional consequences as shown in a previous study. 33 However, further in vitro experiments are needed to prove those hypotheses.
It is noteworthy to mention that our analyses and interpretation were based on the available sequences in the LANL database. In our dataset, the total number of Tat exon 1 sequences under each subtype varies, and the number of sequences for HIV-1 subtypes B and C was greater than for other subtypes. In particular, there were fewer available sequences for subtypes H, J, and K than for other subtypes. However, as mentioned earlier, this variation derives from the global distribution of HIV-1 subtypes; as a result, it influences the available deposited sequences in LANL. For example, there are more deposited sequences for subtypes B and C than for other subtypes as they account for about 60% of the total number of HIV-1 infections worldwide. Since the LANL sequence database is a widely recognized data source for HIV evolutionary studies, we assume that our analyzed data sets are representative and adequate enough for investigating the intersubtype genetic variation of HIV-1 Tat. However, the genetic variability of Tat in some subtypes such as H, J, and K needs to be further clarified in a larger sample size. Future studies can also be done on the molecular dating of HIV-1 Tat to improve our understanding of its evolution.
In conclusion, genetic monitoring of HIV-1 Tat needs to be continued to understand the adaptive evolution of the virus as well as to develop effective Tat-targeted therapeutics and vaccines. As stated in previous reports, 11,27,32,34 updating the genetic variation of Tat would be an important foundation for developing an effective vaccine and intracellular single chain anti-Tat antibodies. Similarly, to develop Tat-targeted therapeutics, monitoring the target region at the amino acid sequence level should be prioritized because any major or minor genetic changes on those targets may lead to therapeutic failure. Taken together, in these ways our study results therefore contribute to controlling the spread of the global HIV-1 epidemic.
Footnotes
Acknowledgments
The authors would like to thank Mrs. Karen Lewis for her careful reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.