
Abstract
Mutations in the cyclophilin A (CypA) binding region in the HIV-1 capsid affect their dependency on the known HIV-1 cofactor CypA and allow escape from the HIV-1 restriction factor Trim5α in human and simian cells. Here we study the effect of these mutations in the CypA binding region of capsid on cofactor binding, capsid destabilization, and viral replication in primary cells. We showed that the viral capsid with mutations in the CypA binding region (CypA-BR) interacted efficiently with CypA, but had an increased stability upon infection as compared to the wild-type capsid. Interestingly, the wild-type virus was able to infect monocyte-derived macrophages (MDM) more efficiently as compared to the CypA-BR mutant variant. The lower infectivity of the CypA-BR mutant virus in MDM was associated with lower levels of reverse transcription products. Similar to the wild-type virus, the CypA-BR mutant variant was unable to induce a strong innate response in primary macrophages. These data demonstrate that mutations in the CypA binding site of the capsid resulted in higher capsid stability and hampered infectivity in macrophages.
Introduction
C
Here we studied whether mutations on the CypA binding region affect viral properties such as capsid stability, replication kinetics in different target cells, and innate recognition. We observed that mutations on the CypA binding region reduce capsid affinity to CypA, resulting in a capsid that is less sensitive to CypA-mediated destabilization and proteosomal degradation. Mutations in the CypA binding region did not affect viral replication in activated T cells, however, these mutations did impair infectivity in primary macrophages. Viral infectivity in primary macrophages was restricted at the level of reverse transcription, which indicates that capsid stability is an important factor in this process.
Materials and Methods
Cell isolation and cultures
Peripheral blood mononuclear cells (PBMCs) and monocytes were isolated from buffy coats of healthy blood donors. Written informed consents were obtained from all donors in accordance with the ethical principles set out in the declaration of Helsinki. This study was approved by the Medical Ethics Committee of the Academic Medical Center and the Ethics Advisory Body of the Sanquin Blood Supply Foundation in Amsterdam, The Netherlands.
PBMCs were isolated from buffy coats using Lymphoprep (Axis-Shield, Oslo, Norway) density gradient centrifugation, pooled and cryopreserved. After thawing, the PBMC pool was stimulated for 3 days in Iscoves' modified Dulbecco's medium (IMDM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, 5 μg/ml ciproxin (Bayer, Mijdrecht, The Netherlands), and 1 μg/ml phytohemagglutinin (PHA; Remel Europe, Dartford, UK) at a cell density of 5 × 106 cells/ml in a humidified 10% CO2 incubator at 37°C. PBMC cultures were continued in IMDM supplemented with 10% FCS, 20 U/ml recombinant interleukin 2 (rIL-2; Chiron Benelux, Amsterdam, The Netherlands), 5 μg/ml polybrene (Sigma, Zwijndrecht, The Netherlands), 100 U/ml penicillin, and 100 μg/ml streptomycin at a cell density of 1 × 106/ml in a humidified 10% CO2 incubator at 37°C.
Monocytes were isolated by adherence to plastic and cultured in IMDM (Lonza, Basel, Switzerland) supplemented with 10% (v/v) heat-inactivated human polled serum (HPS), penicillin (100 U/ml; Invitrogen, Carlsbad, CA), streptomycin (100 mg/ml; Invitrogen), and ciproxin (5 mg/ml; Bayer, Leverkusen, Germany) for 5 days at 37°C in a humidified atmosphere supplemented with 5% CO2. 293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C humidified atmosphere supplemented with 10% CO2. HeLa cells (ATCC) were cultured in IMDM supplemented with 10% FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C humidified atmosphere supplemented with 10% CO2.
Virus productions
HIV-1-based lentiviruses expressing the green fluorescent protein (GFP, YFP, and CFP) were produced by calcium phosphate transfection of 293T cells with the lentiviral vector (LV) construct, the packaging construct for gag/pol, vesicular stomatitis virus glycoprotein envelope (pCMV-VSV-G), and rev (pRSV-rev) as described previously. 18 Infectious LVs were harvested at 48 and 72 h after transfection and filtered through a 0.22-μm filter. The LVs were concentrated by ultracentrifugation (2 h at 46,000 × g). Virus titers were quantified by determination of the 50% tissue culture infectious dose (TCID50) on 293T cells. After 24 h, the infected cells were analyzed by FACS. The CypA-BR mutant HIV-1-based LV has been described previously 6 and contains three amino acid substitutions in the CypA binding region (H87Q, A88P, and I91V) of the packaging construct.
For the production of replicating NL4-3 Bal clones, the full-length HIV-1 constructs (containing wild-type and CypA-BR capsids) were transfected into 293T cells using the calcium phosphate method. Viruses were harvested at 48 and 72 h after transfection and filtered through a 0.22-μm filter. Virus titers were quantified by determination of the TCID50 on luciferase reporter TZMbl cells. After 48 h, luciferase substrate [0.83 mM ATP, 0.83 mM d-luciferin (Duchefa Biochemi BV), 18.7 mM MgCl2, 0.78 μM Na2H2P2O7, 38.9 mM Tris-HCl, pH 7.8, 0.39% glycerol, 0.03% Triton X-100, and 2.6 μM dithiothreitol] was added and luminescence was measured for 1 s per well using the Centro LB 960 (Berthold Technologies).
VSV-G pseudotyped single round luciferase viruses were produced by cotransfection of pNL4-3.Luc.R-E- constructs containing wild-type or the CypA-BR mutant capsids (which were placed in the pNL4-3.Luc.R-E- construct by site-directed mutagenesis) or N74D mutant capsid (a kind gift from Dr. V. KewalRamani) and the pCMV-VSV-G in 293T cells using the calcium phosphate method. Infectious viruses were harvested at 48 and 72 h after transfection and filtered through a 0.22-μm filter. Virus titers were quantified by determination of the 50% tissue culture infectious dose (TCID50) on 293T cells. After 48 h, infected cells were washed with phosphate-buffered saline (PBS), luciferase substrate was added, and luminescence was measured.
To determine the ability of HIV-1 virions to incorporate CypA, HIV-1-based lentiviruses were produced by calcium phosphate transfection of 293T cells with the LV construct carrying the CAG promoter and GFP, 19 the packaging construct for gag/pol carrying the wild-type or CypA-BR mutant capsid, pCMV-VSV-G, and rev (pRSV-rev) as described previously, 18 together with pcDNA-CMV-CyPA-myc. The transfection was done in the absence or presence of 5 μg/ml CsA. Infectious LVs were harvested at 48 and 72 h after transfection and filtered through a 0.22-μm filter. The LVs were purified through a sucrose gradient, and further pelleted by ultracentrifugation (2 h at 21,000 rpm in the SW28 rotor). The pellet was dissolved in 65 μl whole cell lysis buffer and used for Western blot. Specific CypA proteins were visualized using monoclonal mouse antibodies against myc-tagged proteins (Invitrogen) and capsid proteins were visualized with anti-p24 antibodies (Moab14). 20
Infections
Unstimulated PBMCs (5 × 166) were inoculated with LV-WT-YFP or LV-CypA-BR mutant-CFP (MOI = 10) for 24 h in a 37°C shaking water bath; subsequently the cells were seeded in a 96 round bottom plate with 2 × 105 cells/well and stimulated with PHA. Expressions of surface markers were analyzed 5 days postinfection by FACS CANTO (BD Biosciences) after staining with fluorochrome-conjugated CD4 PerCP, CD45RO APC, CD38 AP, and CD69 APC, obtained from BD Biosciences. Data were analyzed with FlowJo (Tree Star, Inc.).
To determine the viral replication rates of HIV-1 NL4-3 Ba-L containing the wild-type or CypA-BR mutant capsids, 2 × 106 PHA-stimulated pooled PBMCs were inoculated with 100 TCID50 per virus variant for 2 h at 37°C in a shaking water bath in a total volume of 1.5 ml. Subsequently, the inoculated PBMCs were washed with 5 ml of monocyte-derived macrophages (MDM) supplemented with 10% FCS, and cultured in IMDM supplemented with 10% FCS, 20 U/ml rIL-2, 5 μg/ml polybrene, 100 U/ml penicillin, and 100 μg/ml streptomycin (PBL medium) at a cell density of 1 × 106 per ml in a humidified 10% CO2 incubator at 37°C. On days 5, 8, 11, and 14 after inoculation, 1 × 106 fresh PHA-stimulated pooled PBMCs in 1 ml of PBL medium were added to the cultures. Samples for determination of p24 antigen production were harvested every day after inoculation. All samples were tested for p24 antigen production simultaneously at the end of the experiment using an in-house p24 ELISA. 20
For infection with HIV-1 VSV-G pseudotyped NL4-3 luciferase containing wild-type or CypA-BR mutant capsids, MDM were cultured in 24-well plates (Nunc, Langenselbold, Germany) at a density of 5 × 105 cells per well, and inoculated at day 5 with an MOI of 0.02. After 24, 48, and 120 h, the cells were harvested in TriPure reagent (Roche) for further RNA purification. After 24 h the cells were harvested in L6 buffer for further DNA purification. 21 293T cells and HeLa cells were plated at a cell density of 4 × 103 cells per well and cultured for 24 h. The cells were then inoculated with 1,000 TCID50 VSV-G pseudotyped single round luciferase virus and 48 h later the infectivity of the virus was analyzed by luciferase assay. Luciferase activity was compared using the Student's t-test. During and after inoculation, the cells were treated with different compounds as indicated: MG132 (0.1 and 0.25 μM) and CsA (1 μg/ml).
Plasmid construction
For in vitro production of HIV-1 capsid–nucleocapsid (CA/NC) proteins, CA/NC genes of the wild-type, and CypA-BR mutant packaging constructs were amplified by PCR (primers: CA/NC Fw: 5′-CAT AAT CAT AAT GCC TAT AGT GCA GAA CAT CC-3′ and CA/NC Rv: 5′-GTA TCG AGG TAC CGG ACG AGG GGT CGC TGC C-3′) and cloned into a pET43.1 expression vector.
In vitro production of HIV-1 CA/NC complexes
The HIV-1 CA/NC protein was expressed, purified, and assembled as previously described.
22
Escherichia coli BL21(DE3)pLysS cells (Stratagene) were transformed with the pET43.1 expressing wild-type and CypA-BR mutant CA/NC vectors and grown to OD600 of ∼0.6. Isopropyl β-
CA/NC complexes were assembled in the presence of viral RNA that contains a packaging signal. Viral RNA was transcribed from an EcoRI-digested lentiviral construct 23 in a total volume of 100 μl by T3 RNA polymerase according to the manufacturer's recommendation (Promega). For CA/NC assembly, 100 μg CA/NC was combined with 100 μl of the produced viral RNA, adjusted to 0.5 M NaCl to a total volume of 1 ml, and allowed to assemble overnight at 4°C.
Capsid pull-down assay and Western blot
Assembled CA/NC complexes were coupled to cyanogen bromide (CNBr)-activated Sepharose beads according to the manufacturer's recommendations (Amersham). Subsequently, the beads were washed three times with binding buffer (0.1 M acetic acid, pH 4.0, containing 0.5 M NaCl) followed by a wash with 0.1 M Tris-HCl, pH 8, containing 0.5 M NaCl. The beads were resuspended in 150 μl 0.1 M Tris-HCl, pH 8, containing 0.5 M NaCl and 50 μl of beads was used to pull down cellular proteins from cell lysates.
Cell lysates were prepared from 293T cells using NP40 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% sodium deoxycholate) containing protease inhibitor cocktail (Roche). Cell lysates were used for Western blot analysis or in pull-down assays with in vitro-produced CA/NC particles. Cell lysates and precipitates were resolved by SDS-PAGE and blotted on polyvinylideenfluoride membranes. Specific proteins were visualized using monoclonal mouse antibodies recognizing HSV-tagged proteins (Novagen) and myc-tagged proteins (Invitrogen). Endogenous CypA was visualized using a polyclonal rabbit antibody that recognizes CypA (Affinity Bio-reagents). Specific recognition of the protein was visualized by horseradish peroxidase-labeled secondary antibody recognizing mouse or rabbit immunoglobulins. Alternatively, IRDye 800CW-labeled secondary antimouse antibody and IRDye 680-labeled antirat secondary antibody were used to detect specific recognition of the proteins. Binding of the labeled antibodies was visualized by the Odyssey Imager (Li-Cor Biosciences).
Fate of capsid assay
The “fate of capsid assay” has been described previously. 24 HeLa cells were inoculated with wild-type or CypA-BR mutant LV for 24 h to ensure substantial entry. Next, the cells were detached and washed three times with ice-cold PBS, and subsequently the cells were lysed in a hypotonic lysis buffer (10 mM Tris-HCl, 10 mM KCl, 1 mM EDTA) for 15 min and a freeze-thawing step. Following brief centrifugation to pellet insoluble material, cleared supernatants were layered on a 50% sucrose cushion and centrifuged at 4°C for 2 h at 25,000 rpm. The supernatant was removed and the pellet was resuspended in 500 μl RIPA buffer. Next, capsid proteins were immune precipitated with anti-p24 antibodies (Moab14), 20 washed three times with RIPA buffer, and analyzed by polyacrylamide electrophoresis and Western blotting.
Quantitative PCR
Viral reverse transcription (RT) products were measured with real time quantitative polymerase chain reaction (qPCR). Total DNA was isolated from MDM inoculated with NL4-3 Ba-L 24 h postinfection using L6 buffer [20 g of guanidine isothiocyanate in 100 ml 0.1 M Tris hydrochloride (pH 6.4), 22 ml 0.2 M EDTA (pH 8.0), and 2.5 g Triton X-100] as described previously. 21 Late RT products and integrated DNA were measured using primers and probes detecting Pol and R/U5 products, respectively, 21,25 and Roche Probes and Master mix (Roche, Basel, Switzerland). The following program was used in the LightCycler 480 Real-Time PCR System (Roche): denaturation: 95°C for 10 s; amplification: 55 cycles of 95°C for 15 s and 60°C for 60 s. HIV-1 DNA standards were prepared by serial dilutions of 8E5 cells, which carry a single proviral DNA copy in each cell in uninfected PBMCs. 26
Expression levels of interferon (IFN)-β were measured using RT-qPCR. RNA was isolated from 5 day MDM using TriPure Isolation Reagent (Roche). For quantification of IFN-β, previously described qPCR primers were used. 27 The qPCR was performed with a Lightcycler 480 using SYBR Green I Master (Roche). The following cycling conditions were used: denaturation: 95°C for 10 min; amplification: 50 cycles of 95°C for 10 s, 58°C for 20 s, and 72°C for 30 s. Purity of the PCR products was confirmed by melting curve analysis. IFN-β messenger RNA levels are reported relative to β-actin. Gene expression values were obtained using Roche's Lightcycler relative quantification software (version 1.5.0).
Results
Mutations in the CypA binding region of the viral capsid do not prevent CypA binding
Previously, we demonstrated that three amino acid substitutions (H87Q, A88P, and I91V) in the CypA binding region (CypA-BR) of the viral capsid render virus replication CypA independent and insensitive to human, African green monkey, and rhesus macaque Trim5α-mediated inhibition. 6,13 Furthermore, it was observed that a virus containing mutations in the CypA-BR was unable to saturate the Trim5α-mediated restriction for wild-type HIV-1 in simian cells. 6
To determine whether these mutations in the CypA-BR might influence the interaction between the viral capsid and the cellular proteins CypA, the binding of this cellular protein to in vitro-produced CA/NC 22 containing either the wild-type CypA binding region or a CypA-BR mutant was studied. CA/NC complexes were covalently coupled to CNBr-activated Sepharose and used to pull down CypA from cellular lysates of 293T cells. We observed that in vitro-produced CA/NC containing either the wild-type CypA binding region or a CypA-BR mutant was able to interact with CypA and the amount of CypA bound to CA/NC could be reduced by the addition of CsA (Fig. 1A).
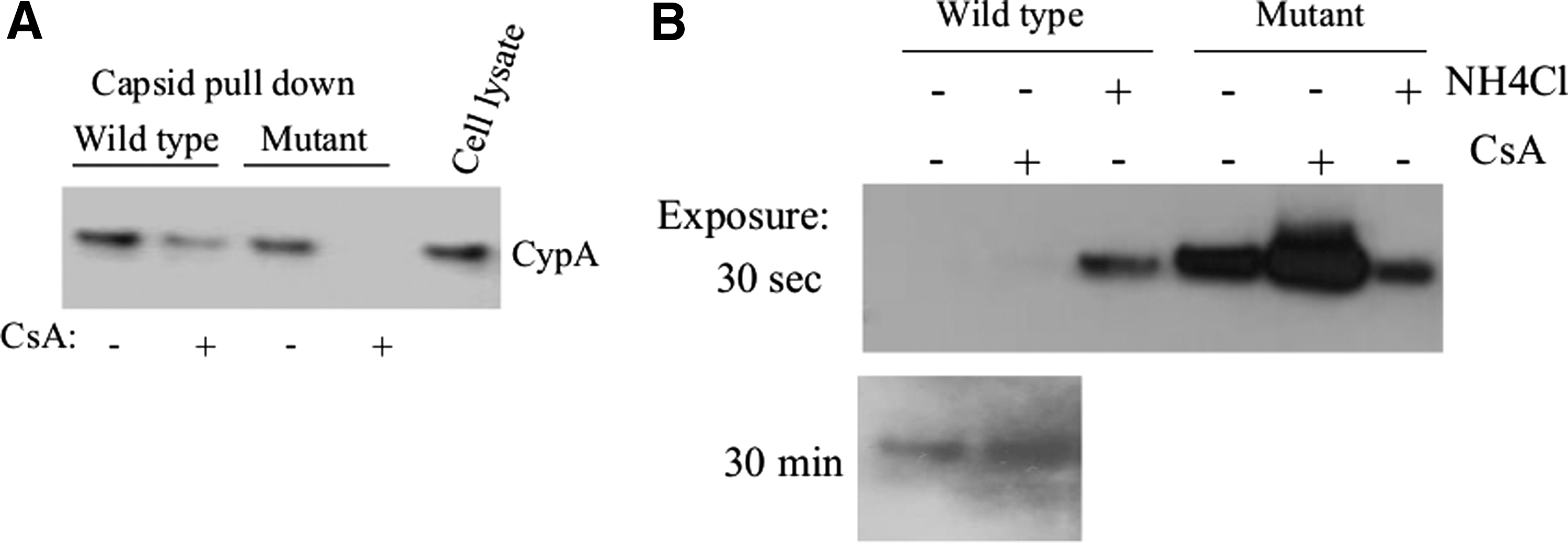
Mutations in the cyclophilin A binding region (CypA-BR) of the viral capsid increase capsid stability.
Furthermore, we analyzed the ability of wild-type as well as CypA-BR mutant capsids to incorporate CypA into the virion. Viral particles containing wild-type capsid or CypA-BR mutant capsid were produced in 293T cells in the presence and absence of CsA. Viral particles were concentrated by ultracentrifugation and purified using a 50% sucrose gradient. In agreement with previous observations, we observed that viral particles containing the CypA-BR mutant capsid encapsidate 30% less CypA into virions as compared to viral particles that contain the wild-type capsid (Supplementary Fig. S1; Supplementary Data are available online at
Mutations in the CypA binding region of the viral capsid increase stability upon infection
The stability of a retroviral capsid after cellular entry is very important for efficient infection. 24,28 Previously, it was demonstrated that binding of CypA to the retroviral capsid strongly induced destabilization of the viral capsid. 29 Here, we analyzed the role of CypA in the destabilization of wild-type and CypA-BR mutant viral capsids using the previously described fate of the capsid assay. 24 HeLa cells were inoculated with VSV-G pseudotyped LV containing the wild-type or CypA-BR mutant capsid (37.5 μg p24 in 15 × 106 cells) in the presence or absence of 1 μg/ml CsA. Twenty-four hours after infection, cells were harvested and washed three times with PBS. Cytosolic extracts were prepared and intracellular capsid particles were pelleted through a 50% sucrose cushion and analyzed by Western blotting. High concentrations of capsid proteins of the CypA-BR mutant LV could be detected in the pellet, whereas only limited amounts of wild-type capsid proteins were detected even after overexposure of the blots (Fig. 1B). This indicated that CypA-BR mutant viral capsids are more resistant to destabilization as compared to wild-type capsids. Moreover, when CsA was added to the cultures during infection, an increased amount of capsid was observed in the pellet fraction (Fig. 1B). These data indicate that mutations in the CypA binding region of capsid can stabilize the viral capsid.
As a control ammonium chloride (30 mM NH4Cl) was added to the cells to inhibit endosome acidification and prevent subsequent entry of the VSV-G pseudotyped HIV-1 into the cytoplasm of the cells thus trapping virus particles in the endosome. FACS analysis of NH4Cl-treated cells indeed demonstrated complete inhibition of infection (data not shown). We observed low amounts of capsid particles in the pelleted NH4Cl-treated cell fractions, which could be the result of endosome rupture during cell lysis. Notably, equal amounts of wild-type and CypA-BR mutant capsid particles were detected in the pellet fraction of the NH4Cl-treated cells, suggesting that the intrinsic stability of the particles does not differ.
Mutations in the CypA binding region make the HIV-1 capsid insensitive to proteosomal degradation
To analyze whether mutations in capsid affect viral infectivity, infection experiments using viruses containing the wild-type or CypA-BR mutant capsid were performed in HeLa cells in the presence or absence of CsA and proteosome inhibitor MG132. HeLa cells were infected with VSV-G pseudotyped NL4-3 luciferase viruses containing wild-type or CypA-BR mutant capsid (1,000 TCID50) for 1 h and cultures were continued in the presence or absence of CsA. MG132 was added to the cultures at different time points: immediately after inoculation (t = 0) and 2, 4, and 8 h after inoculation. The addition of MG132 had only a minor effect on the infectivity of wild-type virus, which indicated that MG132 was unable to rescue viral particles from proteosomal degradation in the presence of high CypA concentrations (Fig. 2).
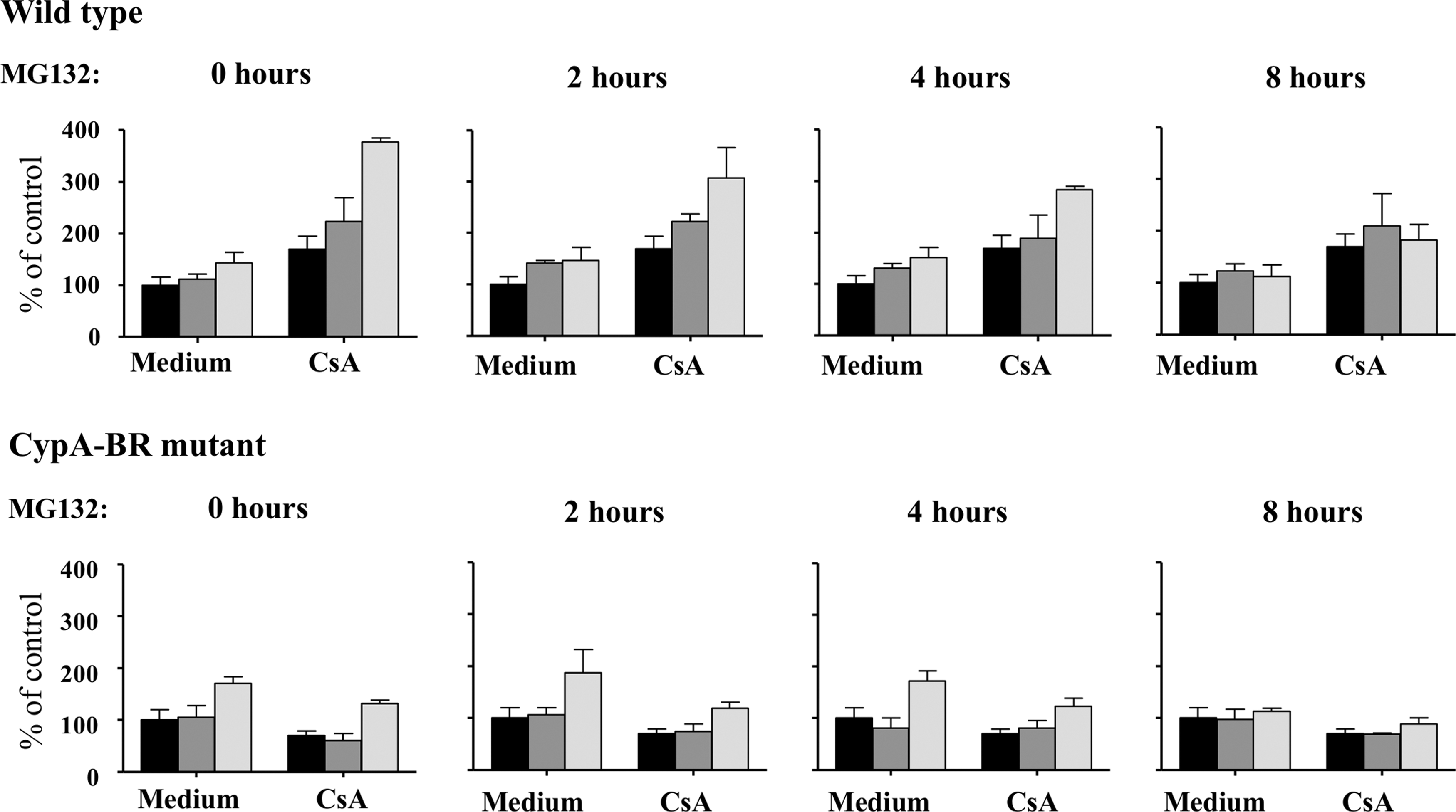
Mutations in the CypA-BR reduce HIV-1 sensitivity to proteosomal degradation. HeLa cells were inoculated with vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped NL4-3 luciferase reporter viruses that contain a wild-type or CypA-BR mutant for 1 h. Cultures were continued in the absence or presence of CsA and MG132 was added at different times after inoculation: 0, 2, 4, and 8 h. At day 2 after inoculation the luciferase activity in the cultures was analyzed. Results are given as mean and standard deviation of triplicates and are representative of three independent experiments. Black bar: untreated control; gray bar: 0.1 μM MG132; light gray bar: 0.25 μM MG132.
However, when CsA was added to the cultures to lower the interaction between the viral capsid and CypA, we observed a strong increase in the infectivity of the wild-type virus in HeLa cells in the presence of MG132 (Fig. 2). This indicates that virus particles are prone to proteosomal degradation shortly after viral entry. Rescue of wild-type virus particles decreased over time, and 8 h after inoculation the addition of CsA and MG132 had almost no effect on the infectivity of the wild-type virus in HeLa cells (Fig. 2). The addition of MG132 had only minor effects on the infectivity of the CypA-BR mutant virus in the presence or absence of CsA (Fig. 2), which indicates that mutations in the CypA binding region make the capsid less sensitive to proteosomal degradation.
Mutations in the CypA binding region of the viral capsid restrict replication in primary macrophages but not in T cells
Next, we examined whether the decreased sensitivity of the CypA-BR mutant virus to capsid destabilization and proteosomal degradation observed in the presence of high CypA also has an effect on viral replication in primary cells. The replication capacity of NL4-3 BAL viruses containing either the wild-type or CypA-BR mutant capsid (100 TCID50) was analyzed on PHA-stimulated PBMCs. Virus replication was analyzed every day for a period of 2 weeks and no differences in the replication kinetics of viruses containing the wild-type or CypA-BR mutant capsid were observed (Fig. 3A). Next we analyzed whether mutations in the viral capsid influence the ability of the virus to infect different cellular subsets within the PBMCs. VSV-G pseudotyped lentiviral vector expressing YFP or CFP containing, respectively, wild-type or CypA-BR mutant capsid proteins was used to inoculate PBMCs. At 24 h postinfection the cells were stimulated with PHA. Five days postinfection the cells were stained for expression of CD4, CD45RO, CD38, and CD69 to determine the amount of infected cells by YFP/CFP expression in naive (CD45RO−) and memory (CD45RO+) CD4+ cells and activated and resting CD4+ cells based on CD38 or CD69 expression. However, we observed no differences in the ability of the wild-type and CypA-BR mutant lentiviral vectors to infect different PBMC subsets (Fig. 3B).

Wild-type and CypA-BR mutant capsid efficiently infect peripheral blood mononuclear cell (PBMC) subsets. Unstimulated PBMCs were inoculated with VSV-G pseudotyped lentiviral vectors LV-WT-YFP or LV-CypA-BR mutant-CFP and were subsequently stimulated with phytohemagglutinin (PHA)
In addition, we analyzed the ability of wild-type and CypA-BR mutant capsids to infect MDM. Monocytes from four different donors were allowed to differentiate into MDM during a 5-day culture and inoculated with wild-type or CypA-BR mutant VSV-G pseudotyped NL4-3 luciferase viruses (MOI = 0.02). Luciferase activity was analyzed at 2 days after infection. HIV-1 containing the wild-type capsid showed a significantly higher infectivity in MDM as compared to virus containing the CypA-BR mutant capsid (Fig. 4A). To investigate whether the mutations in capsid indeed affect early steps in viral replication, the efficiency of reverse transcription was analyzed at 24 h postinfection by qPCR. Indeed, we observed lower levels of proviral DNA synthesis of the virus containing the CypA-BR mutant capsid in MDM (Fig. 4B). These observations suggest that mutations in the CypA binding region of the HIV-1 capsid hamper infection in primary macrophages indicating that a more stable capsid interferes with the efficiency of early steps in the replication cycle in these cells.
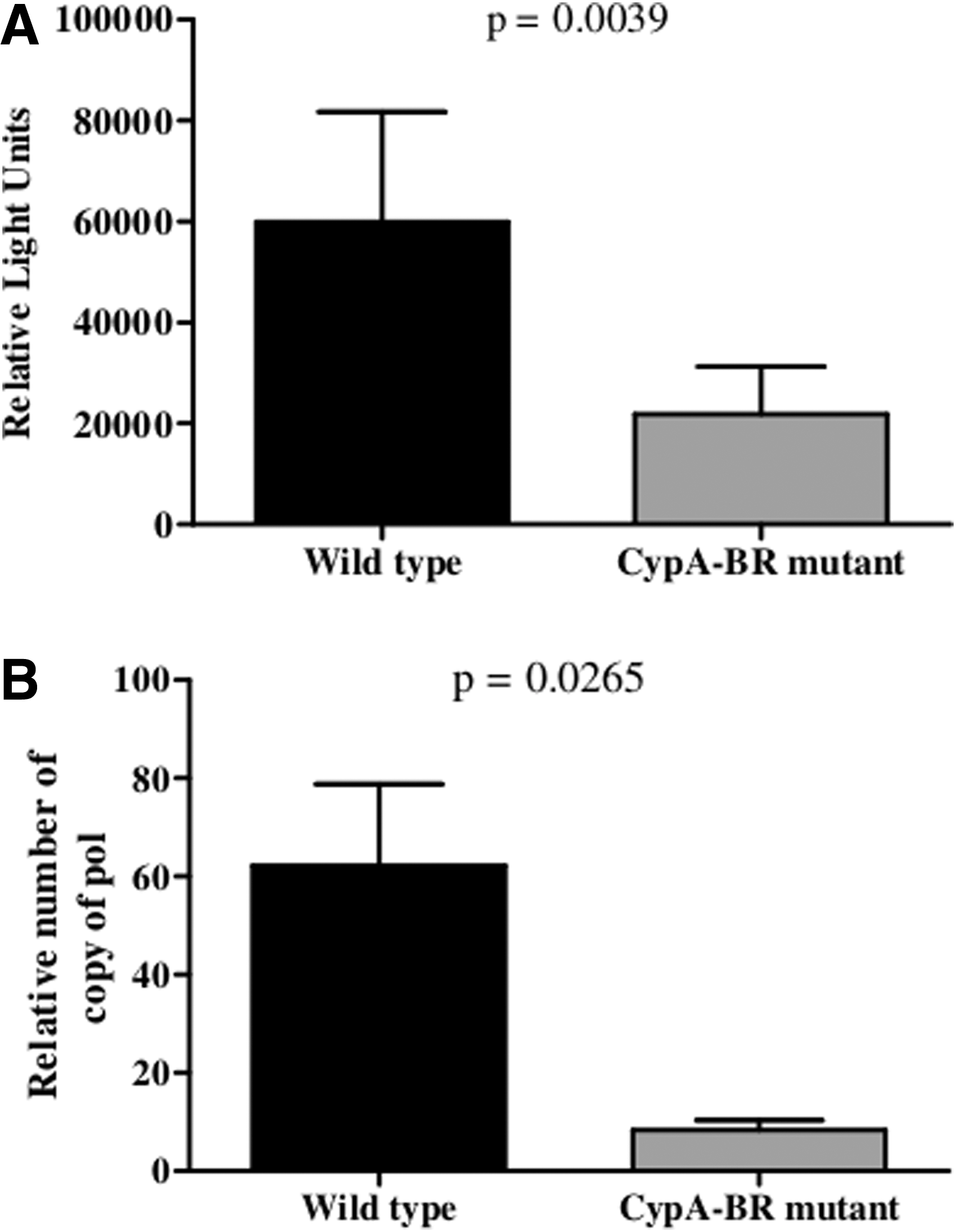
Viral infectivity of the CypA-BR mutant capsid is hampered in primary macrophages. Five day cultured MDM were inoculated with VSV-G pseudotyped NL4-3 luciferase viruses containing wild-type or CypA-BR mutant capsids. Luciferase was measured 2 days postinfection
Mutations in the CypA binding region of the viral capsid do not alter innate signaling in macrophages
It was recently shown that HIV-1 infection in macrophages can activate an innate immune signal, when specific interactions with HIV-1 cofactors (CPSF6) are prevented by the N74D capsid mutation. 30 To determine whether the low infectivity of HIV-1 containing the mutant capsid in macrophages is caused by activation of innate signaling pathways, we analyzed whether viruses containing either a wild-type or a CypA-BR mutant capsid were able to induce an IFN-β response in MDM. HIV-1 capsid mutant N74D, which was previously demonstrated to be insensitive to CPSF631 and induces high levels of IFN-β in MDM, 30 was used as a positive control.
Five day cultured MDM were inoculated with VSV-G pseudotyped NL4-3 luciferase viruses containing wild-type, CypA-BR mutant, or N74D capsid (MOI = 0.02) and mRNA levels of IFN-β were analyzed by qPCR at 24 h, 48 h, and 120 h postinfection. We observed that viruses containing the wild-type or CypA-BR mutant capsid induced only low levels of IFN-β expression (Fig. 5). Confirming previous observations, 30 the HIV-1 variant containing the N74D capsid mutation induced a strong IFN-β response in MDM at 120 h postinoculation (Fig 5). In addition, we observed that the P90A mutant, which cannot bind CypA, 30 is also unable to induce an IFN-B response in macrophages (data not shown). These data indicate that mutations in the CypA binding region of the viral capsid do not affect innate responses in macrophages.

Wild-type and CypA-BR mutant capsids elicit a minimal interferon (IFN)-β response upon infection in primary macrophages. Five day cultured MDM were inoculated with VSV-G pseudotyped NL4-3 luciferase viruses containing wild-type, CypA-BR mutant, or N74D mutant capsids. IFN-β mRNA transcripts were quantified 5 days postinfection. Statistical p values were determined using Student's t-test.
Discussion
Amino acid changes in the CypA binding region of the capsid can alter the phenotype of the virus resulting in CypA-independent infection in human cells and nonrestricted infection in simian cells. 6,13 –15 Here we examined the effects of these mutations on the biological properties of the virus.
We observed that CypA was able to bind efficiently to both wild-type and CypA-BR mutant CA/NC complexes, which could be abrogated with CsA. Similarly, CypA could also be demonstrated in viral particles containing the CypA-BR mutant capsid. This indicates that the CypA-BR mutant capsid does not prevent interaction between CypA and the viral capsid. In HeLa cells, we observed that viruses containing the CypA-BR mutant capsids were abnormally stable since we were able to detect intact capsid particles 24 h after infection whereas the wild-type capsid were degraded at this time. When the interaction between CypA and the viral capsid was abolished by CsA, degradation of viral capsids could be inhibited.
We also showed that infectivity of the wild-type virus can be rescued by the addition of proteosome inhibitor MG132, especially in the presence of CsA, which confirms that high levels of CypA can be detrimental for virus. Indeed, efficient replication of HIV-1 is dependent on CypA levels in the target cell and virus replication is inhibited by both low and high concentrations of CypA. 8,32 –35 CypA-BR mutations in capsid can stabilize the capsid and thereby affect the destabilization process most likely through its altered interaction with CypA. Indeed, MG132 did not have an effect on the infectivity of the CypA-BR mutant virus, confirming that the mutations in the CypA-BR made the viral capsid less sensitive to CypA-mediated destabilization and proteosomal degradation.
In primary macrophages, a significant decrease in replication of the CypA-BR mutant variant was observed in comparison to the wild-type virus. Replication of the CypA-BR mutant was hampered at the level of RT, as demonstrated by the reduced amounts of RT products. We speculated that the reduced interaction between the CypA-BR mutant capsid and CypA slows capsid disassembly thereby limiting access of dNTPs and cellular cofactors into the viral capsid resulting in a decreased rate of RT. However, mutations in the CypA-BR of the viral capsid did not affect viral replication and target cell preference in primary T cells.
Efficient infection of primary T cells requires cellular activation. Activation of the T cells may increase the concentrations of cellular cofactors required for RT and allow for sufficient concentrations of these cofactors even in viral capsids with increased stability. However, it cannot be excluded that the replication of viral variants containing mutations associated with increased capsid stability might be hampered in vivo, due to the lack of cofactors in not fully activated T cells. Mutations in the CypA-BR of the viral capsid have also been observed in naturally occurring HIV-1 variants 16,36,37 and these variants emerged relatively late in infection in a proportion of the HIV-1-infected patients. 16 This indeed indicates that these viral variants do not have a strong selective advantage in vivo.
Mutations in the CypA-BR of the viral capsid have also been associated with viral escape from cytotoxic T cells in HLA-B57-positive patients. 17,37 –40 In particular, H87Q has been demonstrated to be a compensatory mutation for the T110N escape mutation in the HLA-B57-restricted TW10 epitope. 17,36 –38 Here we observed that mutations at amino acid positions 87, 88, and 91 are associated with a more stable capsid. This might indicate that viral escape in the HLA-B57-restricted TW10 epitope results in instability of the viral capsid resulting in attenuated replication. Indeed, viruses that have escaped from the HLA-B57-restricted CTL epitope have increased sensitivity to huTrim5a antiviral activity in comparison to the WT virus. 39,40 Therefore, the emergence of the HLA-B57 escape mutations is accompanied by compensatory mutations 17,36 –38 such as H87Q, which may restore viral replication by stabilization of the viral capsid.
HIV-1 replication in macrophages is dependent on the ability of the virus to evade innate signaling through the interaction with cellular cofactors. 30 We were able to confirm previous observations that the N74D capsid mutation, which abolishes the interaction with the cellular cofactor CPSF6, 31 indeed induced a strong IFN-β response in macrophages. However, the mutations in the CypA-BR that reduced the affinity of the capsid for CypA did not trigger IFN-β production in macrophages. This indicates that decreased viral replication of the CypA-BR mutant in macrophages is not caused by the induction of an innate immune response.
In this study, we observed that mutations at positions H87Q, A88P, and I91V in the viral capsid reduce the affinity to CypA resulting in increased capsid stability. The viral variant with the more stable capsid was hampered in its replication at the level of RT in primary macrophages. The mutations in the CypA-BR did not evoke an innate immune response in macrophages. Therefore it is most likely that the reduced replication in macrophages is caused by limited access to cellular cofactors required for efficient RT as a consequence of the increased capsid stability.
Footnotes
Acknowledgments
This work was supported by the Landsteiner Foundation for Blood Transfusion Research (grant 0435), the Netherlands Organization for Scientific Research (NWO grant 96.36.024), and the Dutch AIDS fund (grants 2004062 and 2010038).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.