
Abstract
A new recombinant form representing a mosaic of HIV-1 subtype B and F1 and designated as CRF42_BF was identified in Luxembourg. We confirmed the inedited nature of CRF42_BF by near full-length genome characterization and retrieved a possible ancestor originating from Brazil. The demographic history of CRF42_BF in Luxembourg using Bayesian coalescent-based methods was investigated. The exponential phase of the logistic growth happened in a very short time period of approximately 5 months associated with a high mean rate of population growth of 15.02 new infections per year. However, CRF42_BF was not characterized by either a higher ex vivo replication capacity in peripheral blood mononuclear cells (PBMCs) or a higher ex vivo transmission efficiency from monocyte-derived dendritic cells to PBMCs as compared to B and F1 viruses. These data do not support a high pathogenic potential of CFR42_BF but rather an initial bursting spread of the recombinant probably due to a more favorable transmission route.
T
Although the global distribution of HIV-1 subtypes is mainly driven by socioeconomic changes and immigration, the regional expansion of certain variants is likely related to virus attributes and opportunity for infection by using a specific route of transmission. 1 It has been assumed that higher or lower fitness and transmissibility may explain the success or failure, respectively, of distinctive subtypes in different regions. 2 Because of its particular geographic location and because of one of the highest net migration rates worldwide, Luxembourg is particularly prone to HIV-1 diversification. 3 Analysis of the B subtype phylogeography revealed that Luxembourg is a migratory target and imports viral diversity from abroad. 4 In this context, surveillance of HIV-1 molecular epidemiology is of high importance and a systematic approach to subtype new HIV-1 viruses in the country was undertaken.
The proportion of non-B viruses in newly diagnosed HIV-1 cases has steadily increased in Luxembourg from 20% to 62% in the past decade. Specifically, newly diagnosed infections with subtype G, C, and CRF02_AG were significantly more frequent due to the natural epidemiological link existing between Luxembourg and Portugal or Cape Verde for subtype G and due to the recent sub-Saharan immigration for subtype C and CRF02_AG. A dramatic increase in the incidence of HIV-1 unique recombinant forms (URFs) has been observed since 2001, with more than 20% of the newly diagnosed patients being infected with URFs. Interestingly, a large part of those recombinant strains was represented by a new B/F1 mosaic virus structure detected in 39 patients since June 2003.
Baseline plasma samples of the first 21 patients infected with a typical B/F1 pol pattern from June 2003 to October 2006 were sequenced on ABIPrism 3130xl, using the BigDye terminator cycle-sequencing chemistry (PE Applied Biosystems, Belgium). To obtain near full-length sequence information, RNA was subjected to multiple polymerase chain reactions (PCRs) generating overlapping subgenomic fragments. PCR products were sequenced on both strands using multiple primers. Sequencing primer sequences and PCR protocols are available on request. The near full consensus sequence of this new strain was obtained by manual concatenating of the partial sequences using the graphic multiple sequence alignment editor SeaView v.3.2. 5
Phylogenetic analysis, using the maximum likelihood approach under a GTR+G substitution model, was applied to the three longest near full-length CRF42_BF sequences (9126–9151 bp) complemented by the Los Alamos National Laboratory (LANL) 2010 subtype reference set. The phylogenetic tree of the three clinical isolates revealed the monophyletic origin of this newly identified CRF, forming a cluster distinct from previously described B/F1 common recombinant forms (Fig. 1). The mosaic structure of the B/F1 recombinant was determined on the consensus sequence of the 21 near full-length sequences with jpHMM. 6 As shown in Fig. 2, the majority of the B/F1 genome is B, with three segments of subtype F1 in the gag–pol region (1 kbp), in the C2–V5 region (0.6 kbp), and in the gp41 ectodomain (0.28 kbp). The new recombinant structure was designated as CRF42_BF in the Los Alamos HIV sequence database.
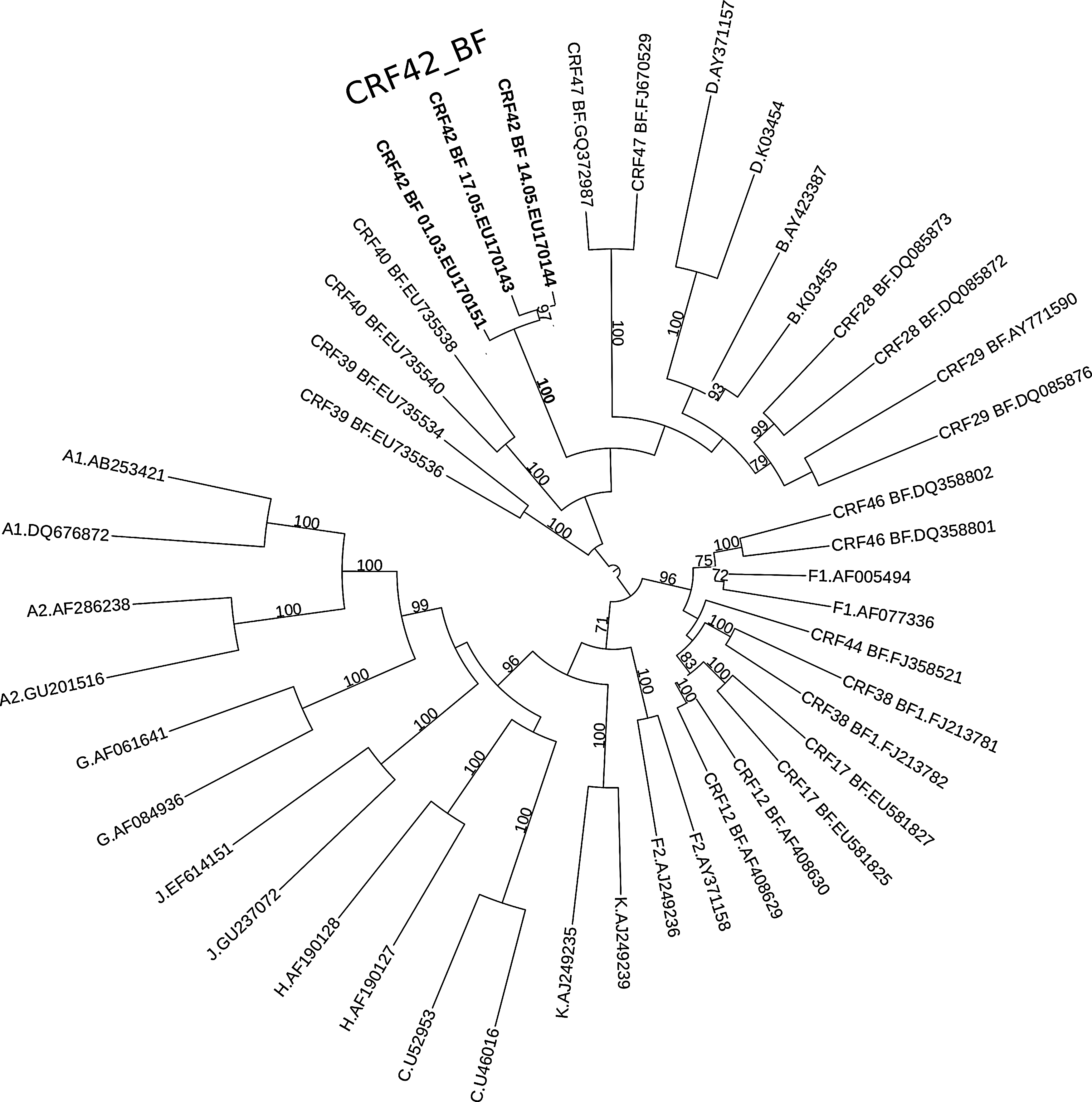
Midpoint rooted phylogenetic tree with the three longest near full-length CRF42_BF sequences (luBF 14.05, luBF 17.05, and luBF 01.03) complemented by two of each pure and B/F CRF subtypes from the 2010 LANL reference set. The maximum likelihood tree was constructed with RAxML 8.0.17 using the GTR+G model. Visualization was done with the online iTOL tool. Values on the branches represent the percentage of the bootstrap analysis performed on 1,000 replicates (only values over 70% are shown). GenBank accession numbers are shown on the right side of the dot symbol for each sequence.
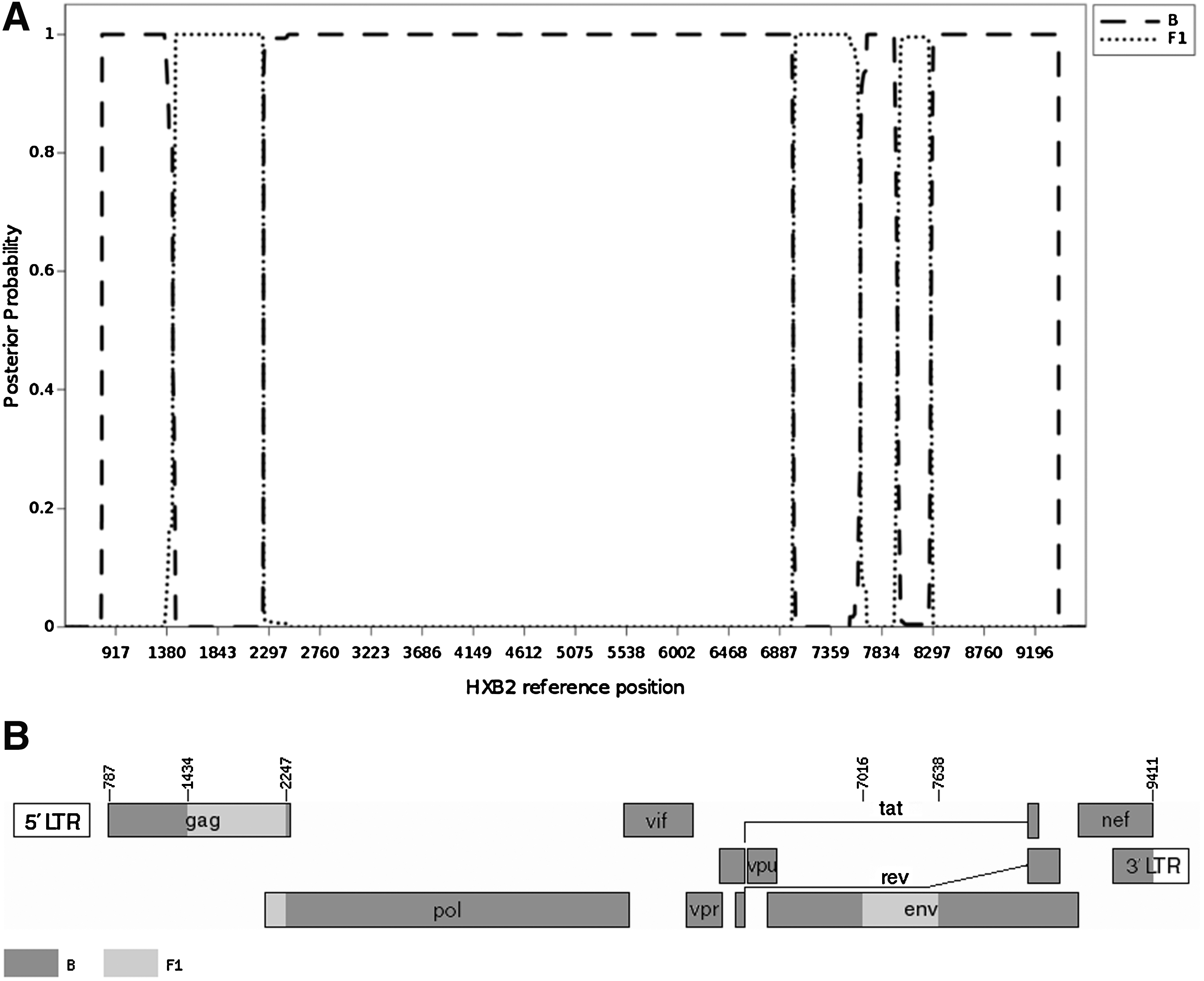
Recombinant analysis of the circulating recombinant form CRF42_BF.
The inedited characteristics of the B/F1 recombinant strains identified in Luxembourg were confirmed using RDP3 7 and Recco 8 showing that the breakpoints identified in the sequences were not identical to those of CRF12, 17, 28, 29, 38–40, 44, 46, and 47_BF (data not shown). The very low genetic diversity observed between the 21 isolates was further suggestive of the explosive nature of this small epidemic, for which a unique introduction event could be assumed. Indeed, the first 12 patients were diagnosed within the first year. Since then the incidence of CRF42_BF cases has been constant, with an average of three new cases per year through December 2013. All patients reported sexual transmission (8 homosexual and 31 heterosexual transmissions). At diagnosis, all the patients presented a mean viral load and CD4 cell count comparable with the mean of the HIV Luxembourg cohort (data not shown).
Most of the CRF42_BF infections were believed to have occurred in Luxembourg rather than having been imported from outside the country. After systematic review of our sequences, we were unable to link the isolated B or F1 segments representing the recombinant to potential parental viruses in Luxembourg. All viral sequences from the European Nucleotide Archive longer than 250 bp (n=1,431,749) were subtyped with COMET 9 to find other potential CRF42_BF sequences. Eleven candidates were further subjected to a bootstrap analysis with the HIV-1 LANL 2010 subtype reference set (1,000 replicates, Jukes–Cantor correction, neighbor-joining trees) and showed at least 97% bootstrap support with CRF42_BF. The earliest sequence, a possible ancestor of CRF42_BF collected during the period of 1986–2001, originates from Brazil, a country characterized by a high frequency of B/F1 recombinants.
As one of the first patients infected with CRF42_BF reported a sexual contact with a partner originating from Brazil in 2003 in Luxembourg, it is tempting to speculate that the CRF was imported from this country. The other 10 sequences originate from different cities in France and were collected between 2006 and 2010. Furthermore, one sequence from Italy was reported to cluster with low bootstrap values with CRF42, indicating other potential introductions of CRF42_BF in Europe. 10 In addition, CRF42_BF was identified between 2005 and 2013 in two individuals from Arlon (Belgium) and four from Nancy (France), two cities bordering Luxembourg. The former sequences were clustered within the sequences from Luxembourg, suggesting the spread of the viruses from Luxembourg (data not shown).
Estimates of the substitution rate and the time of the most recent common ancestor (MRCA) for the 21 near full sequences of CRF42_BF were inferred using a Bayesian Markov Chain Monte Carlo (MCMC) method implemented in the BEAST v1.6.1 program.
11
The adequacy of the convergence was assessed by determining the effective sampling size (ESS) and by visual inspection of the MCMC runs with the Tracer v1.5 program (
Number of substitutions per site per year.
Number of new infections per year.
(Month).
MRCA, most recent common ancestor.
Figure 3 displays the effective number of infections as a function of time for the two datasets: the 21 near full-length genome sequences and the 35 PROT-RT sequences. The effective number of infections represents the number of infections actually contributing to new infections. Both curves show a typical logistic growth with a rapid initial increase and are largely superimposable. When considering only the 21 full-genome sequences, the abrupt increase in effective population size was better ascertained. The exponential phase of the logistic growth happened in a very short time period of approximately 5 months and might be suggestive of a high pathogenic potential of CFR42_BF.
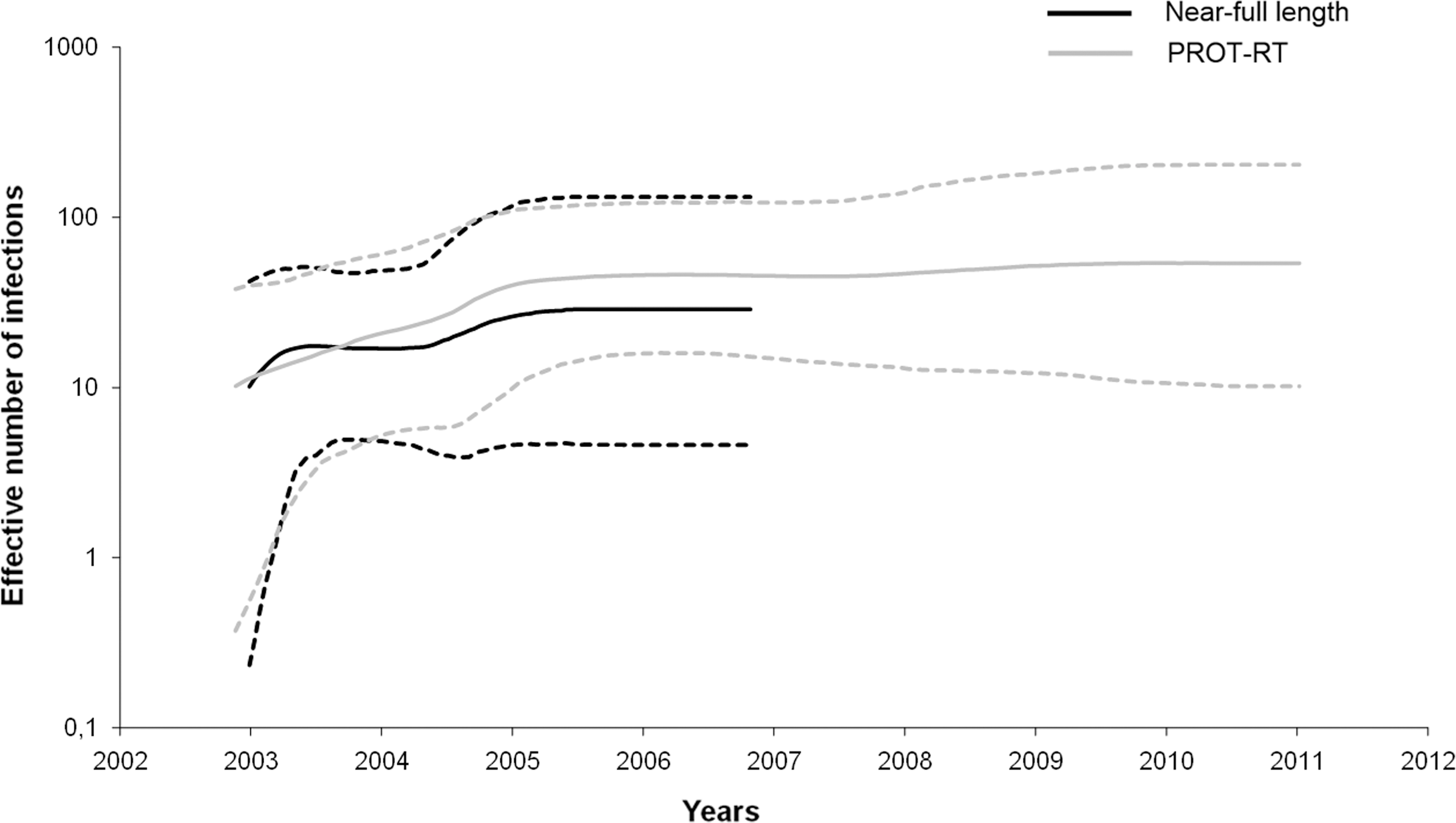
Bayesian skyline plots representing nonparametric estimates of the effective number of infections through time for the CRF42_BF outbreak in Luxembourg, using the 21 near full-length genome (black line) and the 35 PROT-RT genes (gray line). Median estimate of the effective number of infections (solid line) and 95% confidence limits of the estimate (dashed lines) are shown. The vertical axes represent the estimated effective number of infections on a logarithmic scale. The time scale is in calendar years.
B/F recombinants became the predominant genetic forms in Latin American countries where subtype B was predominant before the introduction of subtype F1. It has been proposed that a subtype B and F recombination could provide a higher fitness compared with a pure subtype B and F1 strain. 14 We hypothesized that the high growth rate displayed by the new variant could be related to its fitness in terms of replication and/or transmission capacity. CFR42_BF viruses were strictly R5 variants as determined by their V3 sequences submitted to geno2pheno coreceptor for coreceptor tropism prediction. We therefore investigated the ex vivo replicative capacity of R5-tropic CFR42_BF and phylogenetically related R5-tropic B and F1 subtypes from Luxembourg by selecting 16 patients infected with either CRF42_BF or B and F1 subtypes from 2003 to 2006 at diagnosis with a viral load >20 000 cp/ml.
Frozen virus stocks of six CFR42_BF, six B, and four F1 primary isolates were expanded in short-term cultures of PHA/IL-2-treated PBMCs (Red Cross, Luxembourg) to infect PBMCs and monocyte-derived dendritic cells (MO-DCs). For replication capacity assays, PBMCs were infected and viral replication was monitored for 15 days using p24 concentrations of cell-free viral supernatant measured every 3 days (Perkin-Elmer, United Kingdom). CRF42_BF strains were not characterized by a higher replication capacity as compared to B or F1 viruses, which could explain the rapid spread of the new CRF (p>0.05, data not shown). These results are in agreement with a recent study showing that a similar replicative fitness was shared between subtype B and a B/F recombinant from Argentina. 15
To evaluate transmission efficiency, CD14+ monocytes were isolated from PBMCs by negative selection using the Dynabeads®lMyPure Monocyte Kit 2 (Invitrogen) according to the manufacturer's protocol to achieve a purity of 95% of monocytes. Monocytes were further maturated for 7 days with IL-4 and GMC-SF (20 ng/ml each) to obtain MO-DCs (CD13+ high, CD11c+ high, CD80+ high, CD86+ high, HLA-DR+, and CD83 low), and infected with 0.001 TCID50 of each virus. Autologous frozen PBMCs were added and cultured for 12 days. The p24 antigen was measured at days 5 and 12 and the CRF42_BF viruses did not show a significantly higher in vitro transmission efficiency than B or F1 viruses (p>0.05, data not shown). Consequently, we might assume that CRF42_BF was rapidly introduced in Luxembourg between 2003 and 2006 probably due to a more favorable transmission route rather than by aggressive viral behavior.
Taken together, our population history analysis gives a coherent view of a local epidemic in which the number of cases at least tripled within a few years and remained stable since. We showed that the CRF42_BF outbreak displayed a fast initial growth phase that was not due to superior replicative fitness or transmission efficiency. Ultimately, our study illustrates the increasing molecular complexity of HIV-1 in Luxembourg.
Nucleotide Sequences Accession Numbers
CRF42_BF from Luxembourg: EU170135-155, KJ739817-838. CRF42_BF from Brazil: AY569830. CRF42_BF from France: HQ718234, HQ718277, JQ292572–JQ292577, JN642564. CRF42_BF from Arlon, Belgium: KJ719554-5. CRF42_BF from Nancy, France: KJ739835-8.
Footnotes
Acknowledgments
The study was supported by a grant from the Ministry of Research and “La Fondation Recherche sur le SIDA” of Luxembourg.
Daniel Struck developed the automated tools for the discovery of the new recombinant form, performed the recombination and phylogenetic analyses, and drafted parts of the manuscript. F. Roman discovered the recombinant virus and performed the near full-length genome characterization of CRF42_BF. Sébastien De Landtsheer performed the Bayesian phylogenetic analysis. Christine Lambert, Cécile Masquelier, and Jean-Yves Servais sequenced the near full genome of CRF42_BF. Jean-Yves Servais performed the transmission efficiency assays from Mo-Dcs to PBMCs. Jean-Ruelle and Veronique Venard genotyped the recombinant in Arlon and Nancy, respectively. Monique Nihjuis performed the ex vivo PBMCs replication assay. Jean-Claude Schmit contributed to patient samples collection and participated in the study design. C. Seguin-Devaux conceived the study and drafted the manuscript. All the authors read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.