
Abstract
CRF01_AE, which has led a new epidemic in many provinces in China and has displayed complex characteristics, has now evolved into multiple clusters in China. Some clusters often circulate in specific regions or among specific risk populations in China. To better determine the characteristics of CRF01_AE circulating in Anhui Province, we analyzed CRF01_AE based on gag and pol sequences. Our results showed that CRF01_AE circulating in Anhui Province was clearly divided into three clusters. Cluster 1 covered 90% of the sequences in all CRF01_AE. Among Cluster 1, the sequences from men who have sex with men (MSM) and heterosexuals were interwoven. It is suggested that MSM may play a bridge role in transmitting HIV-1 among the different risk groups.
CRF 01_AE
Anhui Province, located in southeastern China, has more than 35 million people. By October 31, 2014, 11,173 HIV and AIDS patients were reported. Of them, 7,580 were AIDS patients. In the early phase of the presence of HIV in Anhui Province, blood donors were the major transmission route of HIV infection, and HIV subtype B was predominant. 13 Since 2007, sexual transmission has been the major route of transmission, and accounted for 97.0% of all cases in 2014. As the predominant strain circulating in people at risk of sexual transmission, CRF01_AE become the main source of HIV-1. 14
Recently, young people have become the major new group diagnosed as being HIV positive. The age of 77.7% of HIV/AIDS patients ranged from 22 to 49 years. Moreover, people in this age range often became HIV infected through sexual transmission and by transmission by men. The distribution of CRFs or subtypes of HIV changes together with changes in the transmission patterns in a region. Therefore, in this study we analyzed the molecular epidemiological characteristics of CRF01_AE in people ranging in age from 22 to 49 years old.
In total, 416 newly diagnosed HIV-1-positive samples between 2011 and 2013 were collected from Anhui Province. All subjects were diagnosed less than 1 year from their date of infection, and were drug naive at the time of sampling. Following ELISA-positive screening, each patient was confirmed to be infected by Western blot (HIV blot 2.2; Genelab Technologies, Inc., Singapore). Participants did not take antiviral drugs.
Whole blood samples were collected using sterile ethylenediaminetetraacetic acid tubes. The plasma was centrifugally separated at 3,000 rpm within 6 h, then kept at −80°C for viral RNA extraction. The study was reviewed and approved by the ethical committee at the Anhui Center for Disease Control and Prevention. Written informed consent was obtained from all participants after we informed them of the objective of this study.
Viral RNA was extracted from 140 μl of plasma using the QIAamp Viral RNA Mini kit (Qiagen, Valencia, CA). HIV-1 segments gag (p17 and partial p24) and pol (protease and p51RT) were amplified using reverse transcriptase (RT)-nested polymerase chain reaction (PCR). The first PCR reactions were performed using the Superscript TM III one-step RT-PCR system with platinum Taq DNA polymerase (Invitrogen) with outer primer pair GAG-L/GAG-E2 or MAW26/RT21.
The second PCR reactions were performed using the TaKaRa ExTaq kit (TaKaRa Biotechnology Co. Ltd., Dalian, China) with inner primer pair GAGF2/c-gag or PRO-1/RT20. The sequences of primers used in this study have been previously described in detail. 6,15 Amplified PCR products were separated on an agarose gel and purified with the QiaQuick gel extraction kit (Qiagen, Valencia, CA). Purified products were subjected to direct DNA sequencing using an automated ABI 3730/3730xl DNA analyzer by Shanghai GeneCore BioTechnologies Co., Ltd.
To avoid potential laboratory errors, all the nucleotide sequences obtained in this study were screened using the online HIVBLAST Search tool (
Nucleotide sequences were aligned with reference strains using the Clustal W program implemented in MEGA 6.0, and were further adjusted manually. The subtype references and B subtype sequences from other regions of China were retrieved from the HIV-1 database (
As shown in Table 1, the age of most of the newly diagnosed participants ranged from 20∼39 years; this accounts for 68.34% of the participants. Of the participants 95.56% were male. All HIV infection was obtained via sexual transmission. MSM is the predominant route, and accounts for 70.56%. Of those HIV positive 31.11% suffered from AIDS when they were diagnosed.
MSM, men who have sex with men.
From 416 samples, we obtained 380 sequences of gag and 382 of pol. Among these sequences, 207 belong to CRF01_AE, accounting for 54.5%. These were followed by CRF07_BC and subtype B, which accounted for 32.1% (122/380) and 42% (11.1%), respectively. Nine sequences belonged to newly unique recombinant forms (URF), which accounted for 2.3%.
CRF 01_AE was predominant among all HIV-1 circulating in Anhui Province. To confirm the relationship between CRF01_AE circulating in Anhui Province and other provinces in China, we constructed phylogenetic trees of sequences belonging to CRF01_AE along with reference sequences used in previous studies.
As shown in Figs. 1 and 2, three distinct clusters were included in CRF01_AE circulating in Anhui Province. Cluster 4 is the predominant lineage, accounting for more than 90% of sequences. Cluster 2 and Cluster 3 account for 7% and 3% of sequences, respectively. The sequences isolated from Anhui Province account for more than 90% of those within Cluster 4, Cluster 1, and Cluster 2, respectively (Figs. 3 and 4). Cluster 4 corresponds to Cluster 4 in Feng et al. and Cluster 3 in Zeng et al. 7,8 Cluster 1 was corresponds to Cluster 1 and Cluster 5 in Feng et al. and to Cluster 2 and Cluster 4 in Zeng et al. 7,8 Cluster 2 corresponds to Cluster 2 and Cluster 6 in Feng et al. 7
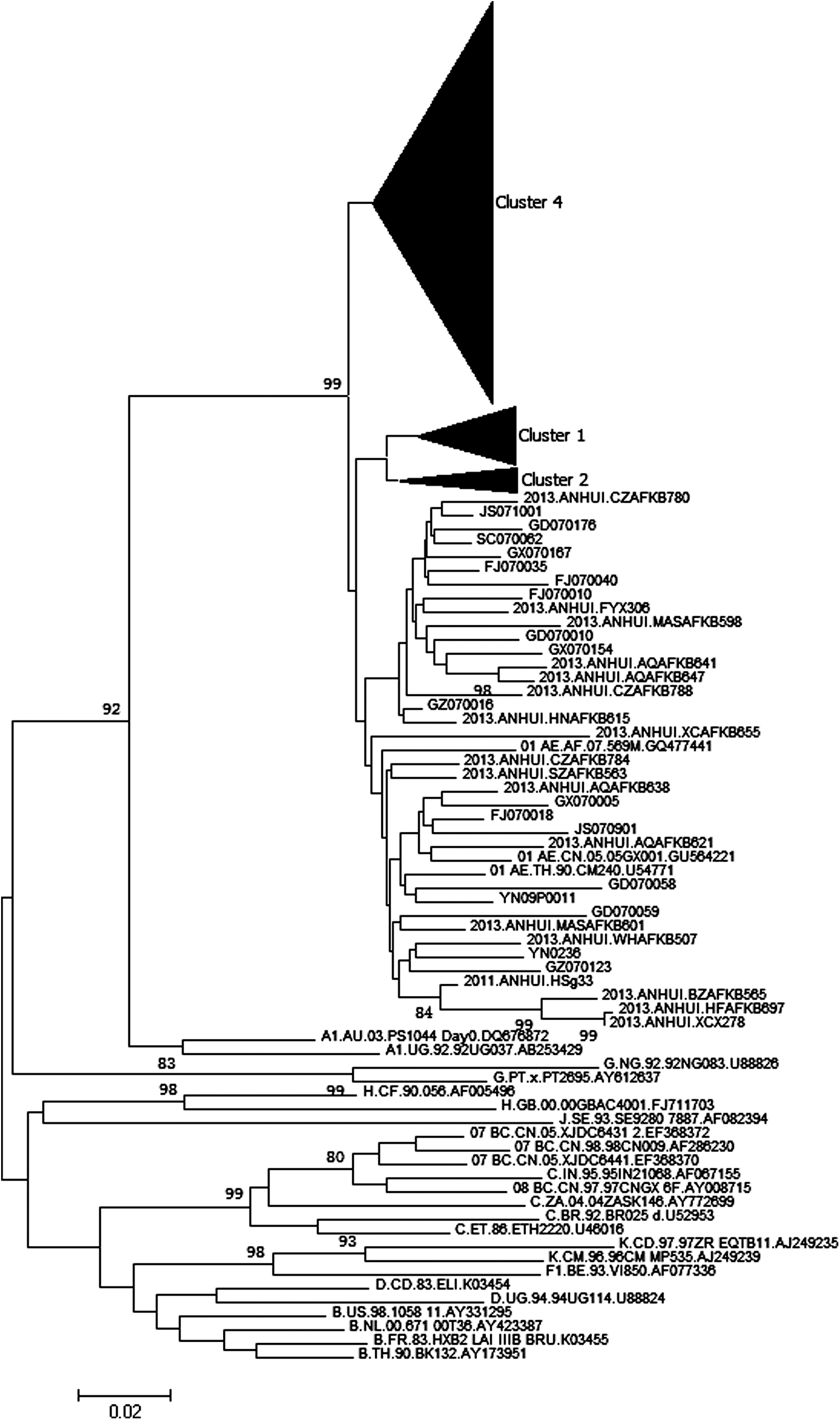
Phylogenetic analysis of gag region sequences. The phylogenetic tree was constructed with MEGA 6.0 using the neighbor-joining method. The stability of the nodes was assessed by bootstrap analysis with 1,000 replications, and only bootstrap values >75 were shown at the corresponding nodes.

Phylogenetic analysis of pol region sequences. The phylogenetic tree was constructed with MEGA 6.0 using the neighbor-joining method. The stability of the nodes was assessed by bootstrap analysis with 1,000 replications, and only bootstrap values >75 were shown at the corresponding nodes.
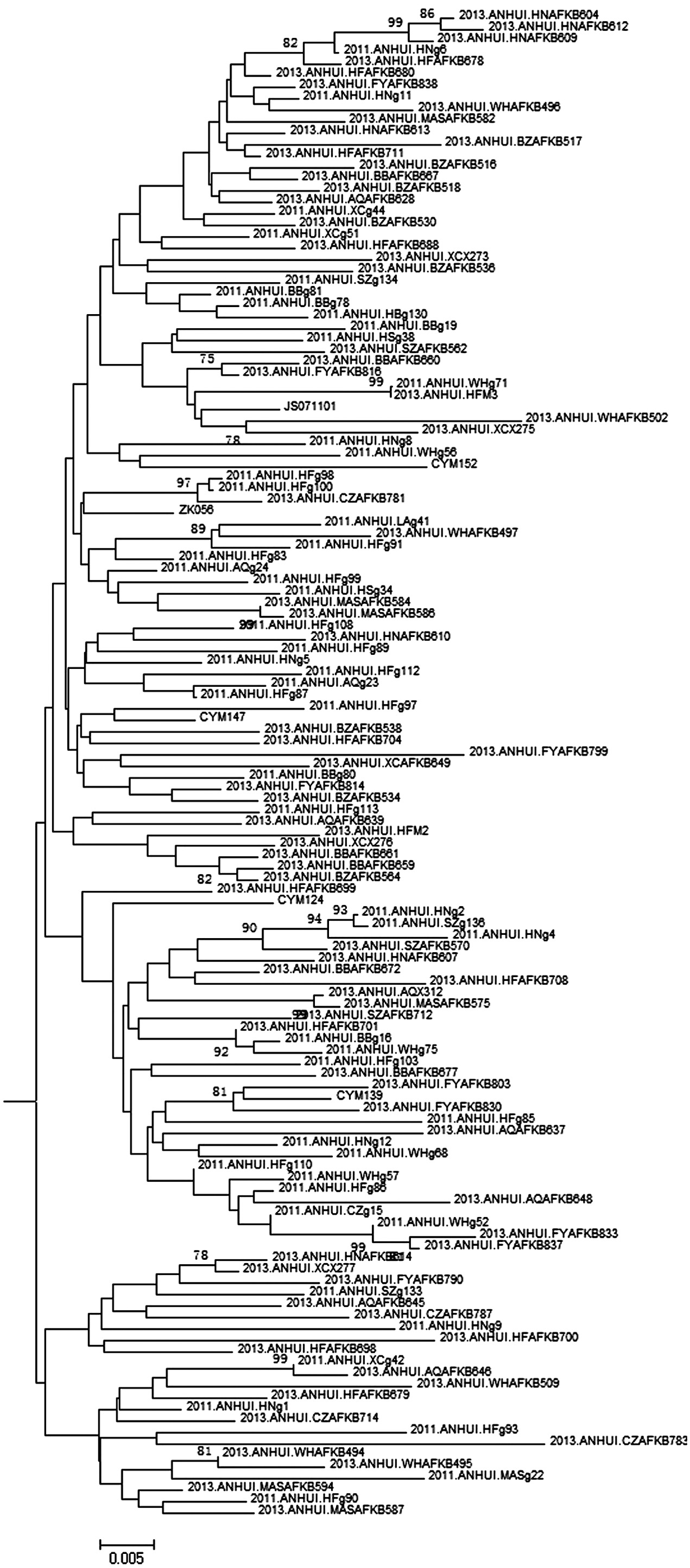
Phylogenetic analysis of cluster 4 in Fig. 1 based on the gag region, which is the phylogenetic tree before it collapsed.
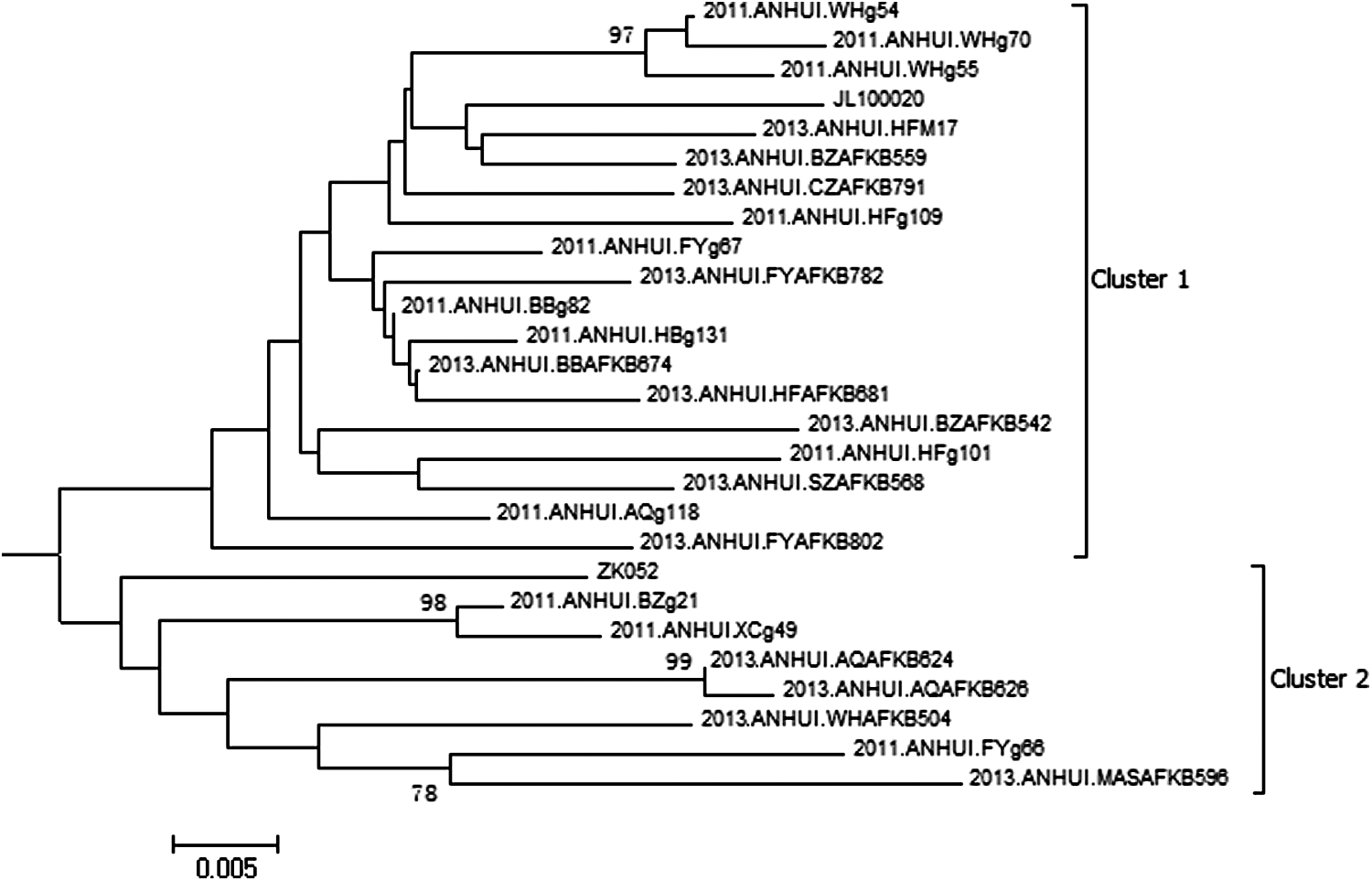
Phylogenetic analysis of clusters 1 and 2 in Fig. 1 based on the gag region, which is the phylogenetic tree before it collapsed.
In previous studies, CRF01_AE circulating in China was divided into multiple clusters. The circulation of the cluster varied based on the specific region and transmission route. As in the study by Feng et al., Cluster 4 was composed of two subclusters, 4a and 4b. 4a was exclusively from MSM in northern China whereas 4b contained CRF01_AE strains from non-MSM populations. However, the predominant Cluster 4 in this study was not divided into two distinctly different subclusters. The sequences corresponding to Cluster 4a and Cluster 4b were intertwined.
In the study by An et al., CRF01_AE circulating among MSM in northern China was composed of four clusters. Our study has shown that CRF01_AE in MSM mingled with those in heterosexuals. These results suggested that the factors affecting HIV transmission are different in various regions of China. In China, the sexual activity of MSM was more complex than in other regions in the world. As we know, MSM living in China not only often have many homosexual partners, but also have heterosexual relations with their own wife or with heterosexual partners or buy commercial heterosexual services. 16 –19 Therefore, MSM play a bridge role in the transmission of HIV between MSM and heterosexuals as well as among heterosexuals in China. 20 The MSM population may make the spread of HIV more complex and increase the challenges of intervening in the transmission of HIV in China.
Sequence Data
The sequences using in this study have been submitted to GenBank with accession number KPKP992167–KP992342, KP992343–KP992441, KC203134–KC203202, and KC203252–KC203325.
Footnotes
Acknowledgment
This work is supported by the National Grand Program on Key Infectious Disease Control (2012ZX10001-002-001-002).
Author Disclosure Statement
No competing financial interests exist.