
Abstract
Approximately 35 million people worldwide are infected with human immunodeficiency virus (HIV) around 3.2 million of whom are children under 15 years. Mother-to-child-transmission (MTCT) of HIV-1 accounts for 90% of all infections in children. Despite great advances in the prevention of MTCT in Brazil, children are still becoming infected. Samples from 19 HIV-1-infected families were collected. DNA was extracted and fragments from gag, pol, and env were amplified and sequenced directly. Phylogenetic reconstruction was performed. Drug resistance analyses were performed in pol and env sequences. We found 82.1% of subtype B and 17.9% of BF recombinants. A prevalence of 43.9% drug resistance-associated mutations in pol sequences was identified. Of the drug-naive children 33.3% presented at least one mutation related to protease inhibitor/nucleoside reverse transcriptase inhibitor/nonnucleoside reverse transcriptase inhibitor (PI/NRTI/NNRTI) resistance. The prevalence of transmitted drug resistance mutations was 4.9%. On env we found a low prevalence of HR1 (4.9%) and HR2 (14.6%) mutations.
G
The defining feature of HIV is its exceptional genetic diversity. The pandemic M group has been classified into nine genetic subtypes: A–D, F–H, J, and K, and recombination between these subtypes has generated more than 70 circulating recombinant forms (CRFs) [
Brazil was the first developing country to implement a countrywide public health program to prevent MTCT, providing pregnant women and newborns with free HIV diagnostic tests and antiretroviral therapy (ART). 2 While ART has been shown to reduce MTCT rates, transmission of resistant strains has also been documented. 5
This cross-sectional study was performed with a sample of 41 HIV-1-infected individuals: children/adolescents infected though MTCT (22) and their mothers (19), followed at the Specialized State Center in Diagnosis, Care and Research (CEDAP), a Reference Health Service located in the city of Salvador, Bahia, Brazil. The study population was consecutively recruited in 2012/2013 and clinical data were obtained from patients' medical records.
The whole blood samples were obtained and genomic DNA was extracted using the Qiagen extraction kit (Qiagen, Valencia, CA). Fragments of gag (positions 836–2040), pol (positions 2253–3260), and env (positions 6817–8296) genes, all three relative to the HXB2 reference sequence, were generated by nested PCR, as previously described. 6,7 The DNA fragments were purified and sequenced in an ABI 3130xl Genetic Analyzer (Applied Biosystems) using a Big Dye Terminator kit (Applied Biosystems).
Three different datasets were created according to each genomic region and each one included 54 subtype-specific reference sequences. The final fragment length and the position relative to HXB2 were gag: (1079 bp, position 895 to 1974), pol (900 bp, position 2358 to 3258), and env (1242 bp, position 6921 to 8163). The alignment of a comprehensive list of reference sequences (A–D, F–H, J, and K) was retrieved from the HIV Los Alamos database (
The sequences of all datasets were aligned and manually edited. The subtype of HIV-1 sequence samples was determined using Rega HIV-1 Subtyping Tool V.3 (
To check the presence of drug resistance-associated mutations (DRAM) in pol (PR/RT fragments), sequences were submitted to the Stanford HIV resistance database (
Of the 41 HIV-1-infected subjects, 22 (53.7%) were vertically infected children/adolescents and 19 (46.3%) were their mothers (adults). Clinical, demographic, and laboratory data of the studied subjects are described in Table 1. Regarding use of ARVs, 7 (31.8%) children and 12 (63.2%) mothers were under HAART, while 15 children (68.2%) and 7 (36.8%) mothers were drug naive. Among seven children treated with ARV, three (42.9%) had received nucleoside analogue reverse transcriptase inhibitors (NRTI) + nonnucleoside analogue reverse transcriptase inhibitors (NNRTI) and four (57.1%) had received an NRTI + a protease inhibitor (PI). Among 12 mothers treated with ARV, half of them received a NRTI + NNRTI and half NRTI + PI. None of them used fusion inhibitors (FI) or CCR5 antagonists. Of the individuals on therapy, four (57.1%) of the children and eight (66.6%) of the mothers had undetectable viral load (i.e., VL < 50 copies/ml).
Three mothers with two offspring.
IQR, interquartile range; ARV, antiretroviral; IV, intravenous; N/A, not applicable.
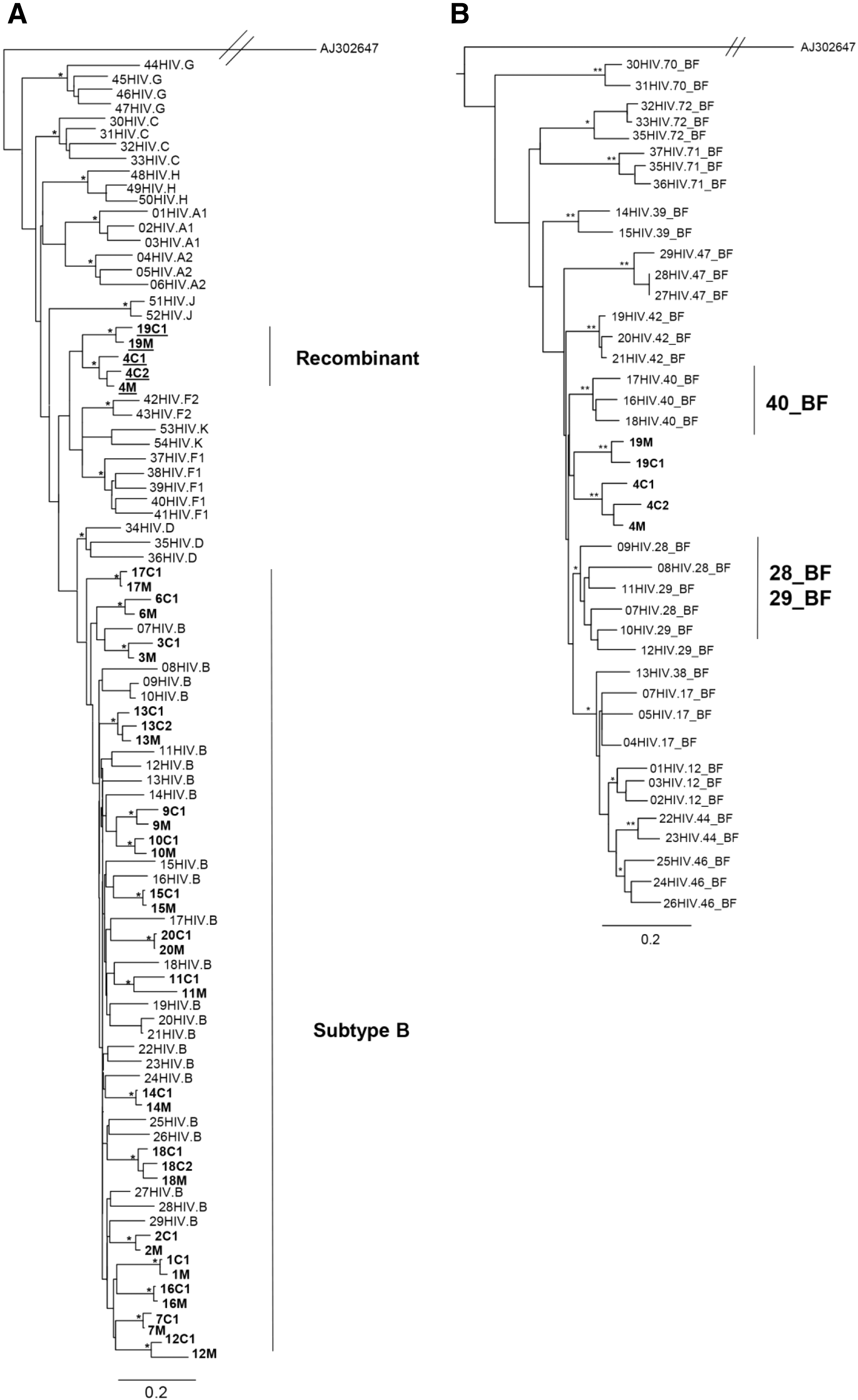
All samples were successfully sequenced in the three genomic regions, except gag sequences from family 8. The ML phylogenetic analyses of the partial gag, pol, and env genes are shown in Figs. 1A, 2A, and 3. Gag and pol phylogenies were inferred with the GTR + I + G model of nucleotide substitution, while env phylogenies were inferred with the HKY + I + G model of nucleotide substitution. In the three trees all mother and child sequences were more closely related to each other than to any other sequences, except pair 6 in the pol ML tree. In this case, the mother–child sequences clustered together in gag and env trees but unexpectedly clustered separately in pol.
Phylogenetic and subtyping analysis of the gag gene.

Phylogenetic and subtyping analysis of the pol gene.

Maximum likelihood tree based on env sequences. Maximum likelihood tree showing the phylogenetic relationship among 41 HIV-1 samples from this study (in bold) and HIV-1 group M reference sequences from different subtypes. The scale bar at the bottom indicates 0.2 nucleotide substitutions per site. The (*) along the branches represents significant statistical support for the clusters subtending that branch (bootstrap ≥ 70%). Different subtypes are indicated by brackets.
The majority of the gag and pol sequences clustered within subtype B. Five (17.9%) gag and seven (17.1%) pol sequences clustered as outliers to the subtype B clade and were identified as recombinant forms. All env sequences clustered within HIV-1 subtype B.
To further characterize the possible recombinant strains circulating in this population, sequences that did not cluster inside any pure subtype clusters were submitted to phylogenetic analysis using some CRFs as reference sequences. Within the gag sequences, families 4 and 19 were characterized as BF recombinant and confirmed with SimPlot analysis. However, these sequences did not cluster with any recombinant profile and formed a monophyletic cluster between CRF28_BF/CRF29_BF and CRF40_BF sequences in the phylogenetic tree (Fig.1B). Within the pol sequences, strains from families 4, 17, and 19 were characterized as BF recombinant. Families 4 and 19 clustered together with CRF29_BF and CRF28_BF sequences, whereas strains from family 17 were closely related to CRF12_BF sequences (Fig. 2B).
Based on the Stanford algorithm, the prevalence of DRAM was investigated in the pol sequences of HIV-1 isolates in this population. Eighteen (43.9%) individuals presented at least one resistance-associated mutation in the reverse transcriptase (RT) and/or in protease (PR) regions. Half of them were drug naive (five children and four mothers) and the other half were ARV treated (two children and seven mothers). Among the samples showing DRAM, six (25%) presented mutations associated with a high level of resistance to ARV drugs. Four were ARV treated and three of them were using at least one of the drugs as part of their current ARV regimen, while one patient presented a mutation associated with a drug that was not part of her antiretroviral combination. The prevalence of DRAM in naive children was 33.3%. The most frequent DRAMs in the general population were PI (minor) A71T/V (9.1% for drug naive; 21.1% for ARV treated); NRTI M184V (4.5% for drug naive; 4.5% for ARV treated); and NNRTI K103N (9.1% for drug naive; 10.5% for ARV treated). When analyzing the 12 subjects with undetectable VL, three (25%) presented clinically relevant mutations and only one of them had DRAMs related to their current ARV treatment.
Based on the CPR algorithm, we observed the presence of tDRM in only one child (4C2—K103N) and in one mother (16M—M41L, M184V, L210W, K103N), corresponding to a prevalence of 4.9%.
Regarding the env gene, the L44M primary mutation was present in two subjects (4.9%) while the N37K compensatory mutation was found in six subjects (14.65%). All subjects harboring HR1 and/or HR2 mutations had no prior exposure to antiretroviral or prophylactic treatment except one (4C1), who was ARV treated.
In this study, we found 82.1% of subtype B and 17.9% of BF recombinants based on the sequences of gag, pol, and env genes. Similar findings were found when more than one genomic region was evaluated. 10 –12 The sequences of all mother–child pairs were in subtype accordance, including those harboring BF recombinant strains. It is likely that the recombinants exhibit some advantages over parental strains with regard to any changes in tropism and viral fitness, making them potentially more virulent and more efficiently transmitted. 13 As expected, sequences from each family/mother–child pair formed a cluster in all the phylogenetic analyses and were more closely related to each other than to any other sequences, showing the closest pairwise relationship, with high bootstrap levels, which is consistent with vertical transmission.
The exception occurred with pair 6 in the pol tree and this can be explained as a case of divergent evolution in the infant following HIV-1 acquisition and ART. The child was 14 years old at sampling time, he was breastfed for 5 years, and he was diagnosed at 6 years of age and then started HAART. It suggests the virus could have evolved significantly in comparison with the strain that was originally transmitted from the mother before initiation and during ART or that the mother was coinfected with another strain, which recombined with the original one, in a way similar to the famous Zambian couple recombination case. 14
Transmitted drug resistance was defined according to the CPR, an algorithm specifically designed for the epidemiologic surveillance of HIV-1 tDRM. 8 The prevalence of tDRM detected in this population was 4.9%, which was somewhat lower than that previously reported for adults and children in other regions of Brazil, which used the Stanford CPR criteria for these estimates. 15 However, if all DRAMs are included, we found a prevalence of 43.9% DRAM in the pol sequences. Of the drug-naive children/adolescents 33.3% (5/15) presented at least one mutation related to PI/NRTI/NNRTI resistance. However, these were minor resistance mutations and their resistance drug profile did not present intermediate or high resistance to any drug.
Resistance mutations associated with fusion inhibitor (T-20) therapy have been described in the 36–45 amino acid heptad repeat region (HR1), and compensatory mutations have been described in the HR2 region of the gp41 env gene. 9 We found a low prevalence of HR1 (4.9%) and HR2 (14.6%) mutations. Similar findings have been shown in a study from Reis et al. in the Central-West Region, Brazil, working with a cohort of pregnant women. 16 The L44M primary mutation, found in two subjects, can lead to a loss of clinical response if combined with other mutations in the HR1 region. This mutation has been described in isolates from T-20 drug-naive patients. 16,17 On the other hand, two subjects with an extensive resistance pattern on the pol region had no T-20 resistance, indicating that they may benefit from the T-20 salvage therapy.
We are aware that our observations on genetic complexity may be limited by the relatively heterogeneous, small sample and fragments analyzed. Another limitation of this study is that direct bulk sequencing and genotyping of HIV-1 might not identify minority variants. Despite these limitations, our results highlight the importance of monitoring the spread of HIV-1 and its transmission through the vertical route, molecular subtypes, and drug resistance among pregnant women and exposed children.
The sequences were reported to GenBank under accession numbers KJ094770–KJ094810, KJ094811–KJ094851, and KJ09852–KJ09890.
Footnotes
Author Disclosure Statement
No competing financial interests exist.