
Abstract
Viral variants that predominate during early infection may exhibit constrained diversity compared with those found during chronic infection and could contain amino acid signature patterns that may enhance transmission, establish productive infection, and influence early events that modulate the infection course. We compared amino acid distributions in 17 patients recently infected with HIV-1C with patients with chronic infection. We found significantly lower entropy in inferred transmitted/founder (t/f) compared with chronic viruses and identified signature patterns in Vif and Vpr from inferred t/f viruses. We investigated sequence evolution longitudinally up to 500 days postseroconversion and compared the impact of selected substitutions on predicted human leukocyte antigen (HLA) binding affinities of published and predicted cytotoxic T-lymphocyte epitopes. Polymorphisms in Vif and Vpr during early infection occurred more frequently at epitope-HLA anchor residues and significantly decreased predicted epitope-HLA binding. Transmission-associated sequence signatures may have implications for novel strategies to prevent HIV-1 transmission.
Introduction
T
Following transmission, within-host immune selection pressures drive HIV-1 evolution. The t/f virus initially rapidly diversifies in a characteristic star-like phylogenetic shape. Following this, cytotoxic T lymphocytes (CTLs) play a critical role in the control of HIV infection and mediate another bottleneck event from which escape variants emerge. 11 Human leukocyte antigen (HLA) class I-mediated CTL responses are important predictors of disease progression 12 that drive virus escape mutations that are highly adapted to the host environment. 4,13 –18 Recent studies employing ultradeep sequencing 3,19 confirmed dramatic shifts in the frequencies of epitope variants during the first weeks of HIV infection.
HIV-1 accessory proteins, Vif, Vpr, and Vpu, modify the host cell environment and disable innate antiretroviral defenses. As such, the transmission bottleneck may select for variants that more readily establish productive infection. However, limited data exist on amino acid signature patterns within viral proteins, particularly in HIV-1 subtype C t/f viruses. 20 Moreover, limited data exist on the timing of escape mutations in HIV-1 subtype C accessory genes. 16,18 In this study, we reconstructed the t/f viruses derived from the earliest available sequences from 17 individuals with acute/early HIV-1C infection. To determine if, and to what extent, Vif, Vpr, and Vpu signature patterns are present in HIV-1C, we compared these t/f viruses to an independent dataset of chronic HIV-1C infection sequences. We also examined longitudinal sequence changes within published and predicted HLA-restricted CTL epitopes in Vif, Vpr, and Vpu among 17 early infected patients up to 500 days postseroconversion (p/s) to investigate early immune escape events.
Materials and Methods
Study population
Patients were enrolled in an HIV-1C primary infection cohort in Botswana, 21,22 and a subset of 17 subjects was selected based on the stage of HIV infection: six acutely infected individuals (patient code A to H) and 11 randomly selected early infections (patient code OC to QR) (Table 1). Acutely infected individuals were identified before seroconversion by a positive HIV-1 reverse transcription polymerase chain reaction test accompanied by negative HIV-1 serology (Fiebig stage II). 23,24 Seroconverters were identified within early stage of HIV-1 infection (Fiebig stages IV–V). The time of seroconversion (Day 0) was estimated as the midpoint between the last seronegative test and the first seropositive test for the acutely infected subjects and by mid-point of the corresponding Fiebig stage for the recently infected subjects. 21 Written informed consent was obtained from all study participants, and ethics approval for this research was obtained from the Human Research Development Committee of the Botswana Ministry of Health and the Office of Human Research Administration at the Harvard School of Public Health.
Viral RNA extraction and single-genome amplification
Viral RNA extraction from plasma was carried out using the QIAamp viral RNA Mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions, followed by single-genome amplification as described previously. 25 Briefly, reverse transcription of viral RNA was performed using SuperScript III (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The single-genome amplification was based on the method of limiting dilutions with minor modifications. 26
Most recent common ancestor reconstruction and phylogenetic relationship of vif, vpr, and vpu sequences
A consensus sequence was determined from the earliest available sample point p/s for each patient to approximate the most recent common ancestor (MRCA). Both DIVEIN
27
(earliest time point sequences using Group M outgroup) and earliest consensus MRCA comparisons were made. To assess intrapatient sequence diversity, maximum likelihood (ML) phylogenetic trees were constructed using PhyML
28
and the HKY85 nucleotide substitution model. The tree was visualized in FigTree v1.1.2 (
Chronic Los Alamos National Laboratory sequences used for comparisons
Single-patient HIV-1 subtype C sequences available in the Los Alamos National Laboratory (LANL) HIV-1 database (
Measurement of amino acid variability: Shannon entropy
Shannon entropy is a simple measure of local variation in DNA and protein sequence alignments that reflects in a single value both the number of variants and their distribution.
30
To calculate the comparative Shannon entropy between the MRCA for Vif, Vpr, and Vpu for each of the 17 patients in our primary infection cohort, and those with chronic HIV-1C infection, we utilized the online tool at the LANL HIV Sequence Database:
Determination of amino acid frequency
Amino acid frequencies for each codon position of Vif, Vpr, and Vpu were calculated using the Viral Epidemiology Signature Pattern Analysis (VESPA) program from the LANL HIV Sequence Database (
Amino acid frequencies in the t/f viruses of our early infection cohort were compared with those in chronically infected HIV-1C patients to identify codons where the consensus (most common) amino acid differed between the early and chronic datasets. In cases where the most common amino acid was the same for both groups, we used an exact binomial test to compare those that differed in frequency by ≥1 standard deviation of the mean frequency for the dominant amino acids across the whole protein. To correct for multiple hypothesis testing, we used the Benjamini–Hochberg procedure 31 to compute false discovery rate-adjusted p-values (q-values) and included all sites with a common major variant across the two groups for purposes of multiplicity adjustment. Sites with unadjusted p-value ≤.05 and differing in frequency by ≥1 standard deviation of the mean frequency for the dominant amino acids were considered possible signatures of transmitted versus chronic infection. These tests were confirmed using R. 32
HLA typing of major histocompatibility complex class I
High-resolution HLA typing was performed for all study subjects as described previously.
33
Briefly, the AlleleSEQR HLA Sequencing-Based Typing kit (Celera, Alameda, CA) was used according to the manufacturer's instructions. Contig assembly and assignment of HLA alleles were implemented by Assign SBT ver. 3.5.1.42 (Conexio Genomics, Applecross, Australia). Ambiguous positions were resolved by resequencing; if this was not possible, ambiguous types were imputed to high-level resolution using a published statistical method (
Analysis of known CTL epitopes
Known HLA class I-restricted epitopes within subtype C Vif, Vpr, and Vpu were retrieved from the LANL Immunology Database (
Prediction of epitope binding affinity
NetMHCpan2.8 was used to predict binding affinities of autologous Vif, Vpr, and Vpu peptides to the corresponding patient's HLA class I molecules (
Briefly, autologous Vif, Vpr, and Vpu sequences were cleaved into all possible peptide combinations of nine amino acids (9-mers) due to the preference of HLA class I molecules for epitopes of this length. For each patient, recurring epitope sequences were excluded to yield only unique peptides. The consensus sequences for Vif, Vpr, and Vpu derived from chronically infected HIV-1C patients were processed in the same manner. The epitope:HLA binding affinity is given as an IC50, the predicted half-maximal concentration of peptide required to bind a given HLA. As such, a low IC50 is indicative of high binding affinity. NetMHCpan defines an IC50 of 500 nM threshold for weak peptide-HLA interactions, which we used as the cutoff to define a putative epitope for a given HLA allele.
GenBank accession numbers
Sequences from our cohort have been assigned GenBank accession numbers JQ895561–JQ896230.
Results
Clinical characteristics and interpatient viral quasispecies diversity over 500 days p/s
Sequences of HIV quasispecies were obtained from 17 individuals with acute/early HIV-1C infection (herein early infection) (Table 1) by single-genome amplification and sequencing. ML phylogenetic trees were constructed to visualize vif, vpr, and vpu intra- and interpatient diversity up to 500 days p/s. Patients harbored phylogenetically distinct HIV-1C sequences, with the exception of individuals, OK and OI, who represented a transmission pair (or chain) as described previously (Fig. 1). 25 The interpatient vpu diversity exceeded that of vif and vpr, which was consistent with previous observations in primary infection. 25,36
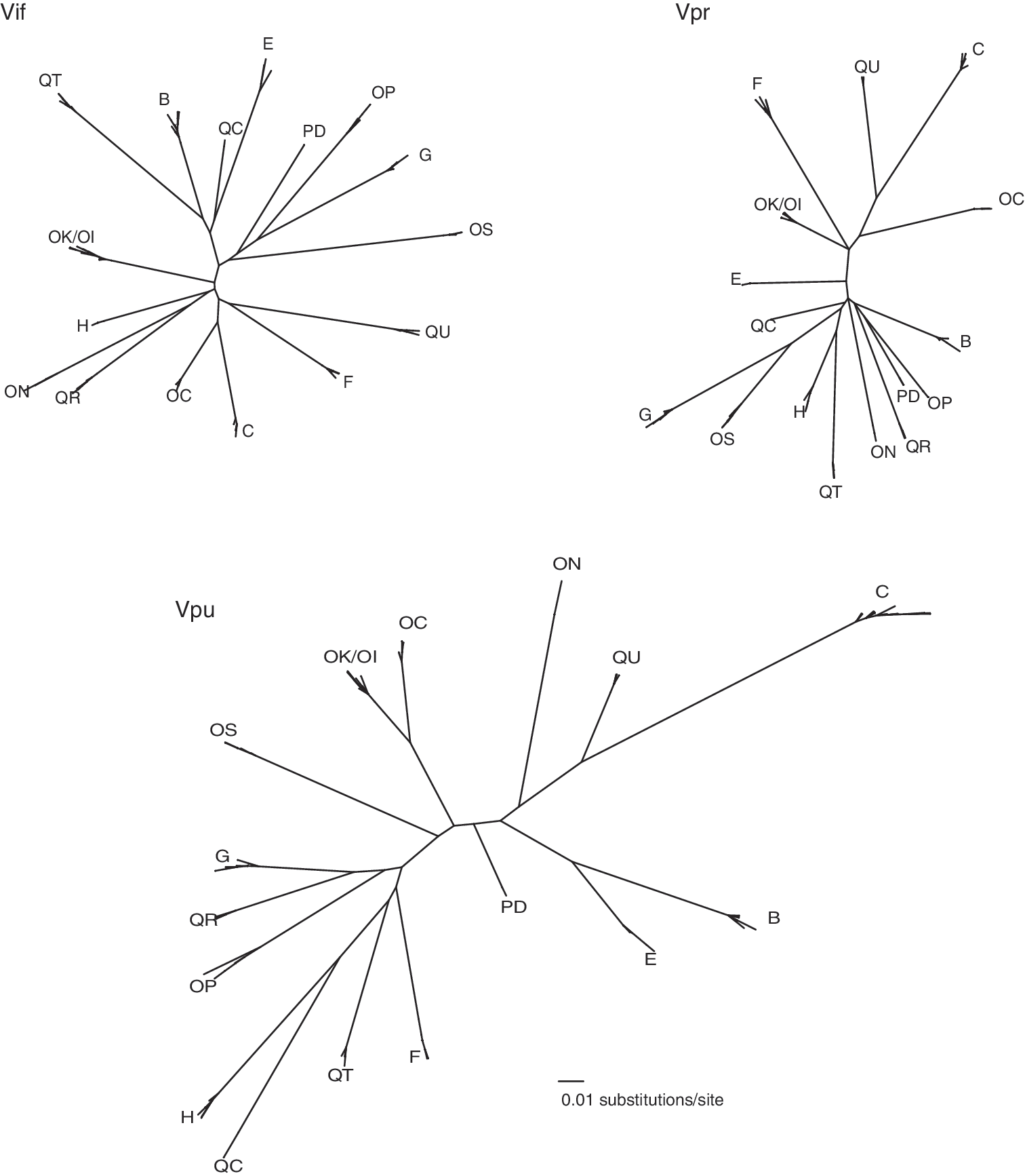
Phylogenetic relationships of vif, vpr, and vpu sequences from 17 individuals during primary infection up to 500 days p/s. Clusters of patient quasispecies are labeled with patient IDs. Phylogenies drawn on the same distance scale highlight interpatient diversity during early HIV-1C infection and greater intrapatient distance for vpu.
Vif and Vpr, but not Vpu, have low entropy in t/f viruses
To investigate HIV-1C Vif, Vpr, and Vpr amino acid diversity, we reconstructed each patient's t/f virus, using the consensus sequence from the earliest time point postinfection, and computed Shannon entropy scores across Vif, Vpr, and Vpu t/f alignments. The median number of sequences per patient used for reconstruction was 10 [interquartile range [(IQR): 5–24]. The reconstructed t/f viruses exhibited relatively low interpatient amino acid entropy in Vif (median 0.00; IQR: 0.00–0.22) and Vpr (median 0.00; IQR: 0.00–0.22) (Fig. 2), suggesting that sequence conservation for these proteins might be important during the establishment of productive HIV-1 infection. In contrast, t/f viruses exhibited higher Vpu entropy (median 0.25; IQR: 0.00–0.65), confirming previous reports of substantial Vpu genetic variability. 37 –39
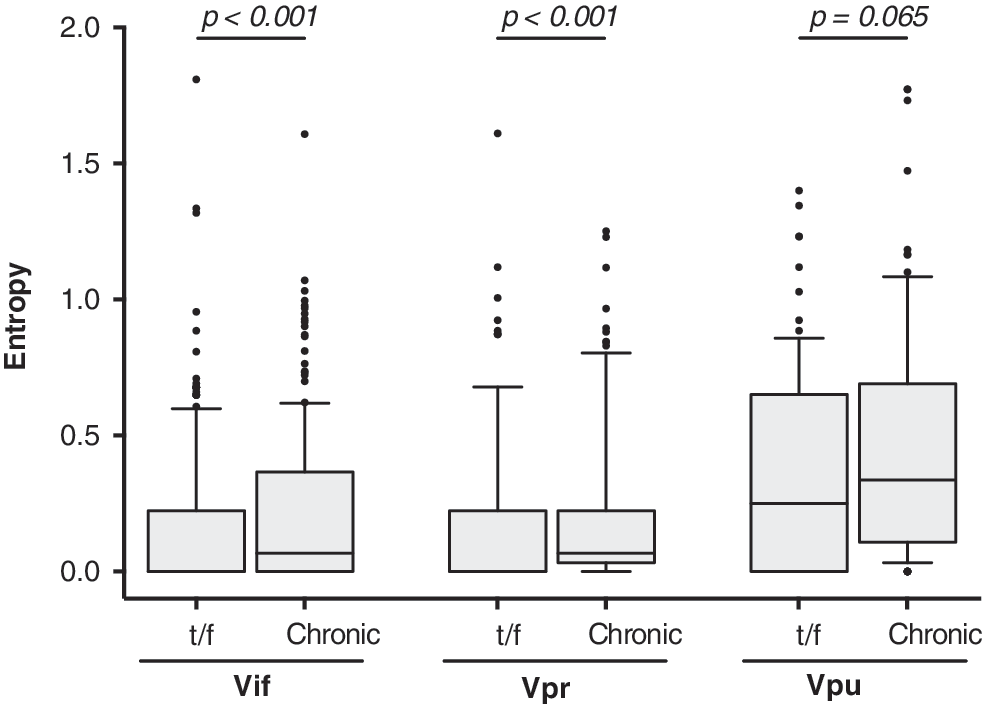
A comparison of Shannon entropy between accessory proteins in t/f viruses compared with chronic infection. Shannon entropy was assessed for full-length protein Vif, Vpr, and Vpu sequences from the t/f viruses of our early infection cohort (n = 17), and HIV-1C chronically infected patients from LANL HIV database (Vif and Vpr n = 187; Vpu n = 197), defined as >1,000 days postinfection/seroconversion. The box-and-whisker plot shows the difference in median entropy with 25%–75%-ile (box) with median (line), 10%–90%-ile (caps), and outliers (black points). t/f, transmitted/founder; LANL, Los Alamos National Laboratory.
As HIV-1C structural proteins generally exhibit lower entropy in early compared with chronic infection, 18 we hypothesized that accessory proteins of t/f viruses would also demonstrate reduced interpatient variability compared with HIV-1C chronically infected patients. As our cohort was only followed up to 500 days p/s, we used HIV-1C sequences from the LANL HIV database (Vif and Vpr n = 187; Vpu n = 197), the majority from Southern African countries (Supplementary Table S1), to represent accessory protein diversity in chronic infection, defined as >1,000 days postinfection/seroconversion. When comparing t/f and chronic Vif amino acid sequences, significantly lower entropy was observed during early infection (0.00 [IQR: 0.00–0.22]) compared with chronic infection (0.07 [IQR: 0.00–0.37]; p < .001, Mann–Whitney test) (Fig. 2). Similarly, entropy of chronic Vpr was higher (median 0.07 [IQR: 0.03–0.22]) compared with t/f viruses (median 0.00 [IQR: 0.00–0.22]; p < .001). Although both t/f and chronic Vpu sequences exhibited substantial variability, there was a trend toward higher entropy in chronic infection (median 0.33 [IQR: 0.11–0.69]) compared with early infection (median 0.25 [IQR: 0.00–0.65]; p = .065).
In t/f viruses, 117/192 (61%) of Vif codons and 62/96 (65%) Vpr codons were completely conserved compared with only 36/82 (44%) of Vpu codons. Regardless of the length of infection (early or chronic), conserved amino acids comprised 41% (78/192) of Vif and 43% (41/96) of Vpr, yet only 20% (17/86) of sites in Vpu. The locations of these conserved residues are highlighted in the context of accessory protein functional domains (Fig. 3).
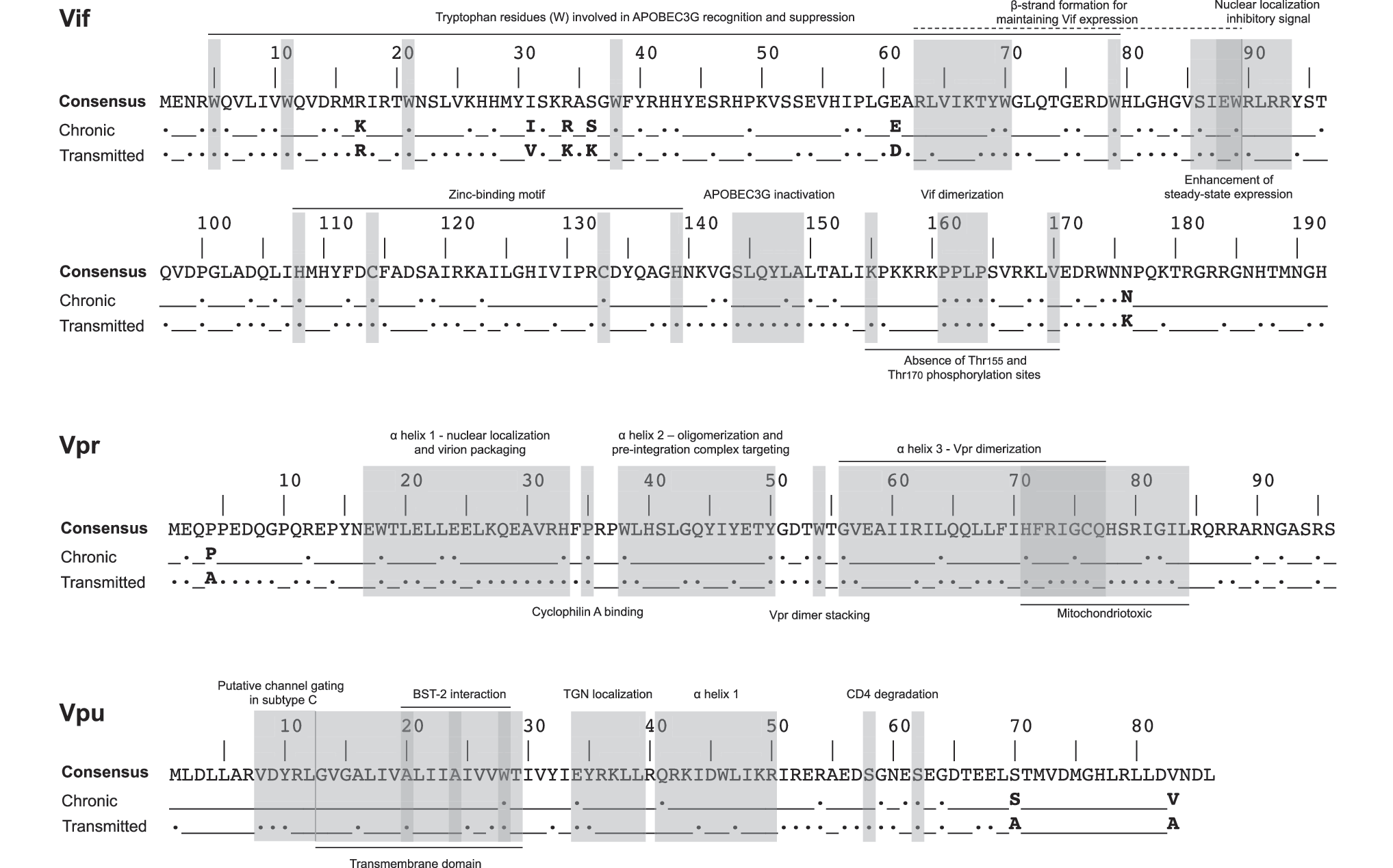
Location of amino acid signature patterns and conservation of functional domain residues in accessory proteins. The HIV-1 subtype C consensus sequence was derived from
Vif, Vpr, and Vpu display distinct amino acid signature patterns in t/f viruses
The significantly lower interpatient amino acid variability in t/f viruses led us to hypothesize that specific genetic transmission signatures might be present within Vif and Vpr, and possibly Vpu. Using the Viral Epidemiology Signature Pattern Analysis (VESPA) program from the LANL HIV Sequence Database, amino acid frequencies in the t/f viruses of our early infection cohort were compared with those in chronically infected HIV-1C patients to identify codons where the consensus (most common) amino acid differed between the early and chronic datasets. 40 In cases where the most common amino acid was the same for both groups, we used an exact binomial test to compare those that differed in frequency by ≥1 standard deviation of the mean frequency for the dominant amino acid across the whole protein (Supplementary Table S2).
Signature patterns are highlighted in Figure 3. In Vif, six codons exhibited a different dominant amino acid in t/f compared with chronic viruses, of which two codons had significantly lower expression of the amino acid most commonly found in chronic infection: I31V (p = .029; q = 0.26) and R34K (p = .004; q = 0.24) (Table 2). We identified an additional 18 codons in Vif whereby the dominant amino acid was the same in the t/f viruses as that in chronic infection, yet differed in frequency by ≥1 standard deviation from the dominant amino acid frequency mean across Vif in chronic infection (Table 3).
t/f, transmitted/founder.
The bold values highlight residues that can more likely be explored as possible transmission signatures that have a p-value < 0.05 and a q-value threshold < 0.3 to correct for multiple hypothesis testing, for which we used the Benjamini-Hochberg procedure to compute false discovery rate-adjusted p-values (q-values).
The bold values highlight residues that can more likely be explored as possible transmission signatures that have a p-value < 0.05 and a q-value threshold < 0.3 to correct for multiple hypothesis testing, for which we used the Benjamini-Hochberg procedure to compute false discovery rate-adjusted p-values (q-values).
In Vpr of the t/f viruses, the frequency of proline at codon 4 was reduced by almost 25% compared with those in chronic infection, although this was not significant (p = .050; q = 0.52). This was the only residue that expressed a different dominant amino acid in t/f compared with chronic viruses (Table 4). Of the t/f residues that differed by ≥1 standard deviation from the frequency of the dominant amino acid in chronic sequences, L22 was significantly enriched in the t/f viruses (p = .007; q = 0.14) compared with chronic sequences, whereas I70 (p = .023; q = 0.28) and A93 (p = .005; q = 0.14) were significantly underrepresented compared with chronic sequences (Table 5). The only residue in Vpu that differed in the dominant amino acid was S70A, although this was not significant (p = .21; q = 1.00) (Table 6), while no Vpu residue differed significantly in the dominant amino acid between t/f and chronic sequences (Table 7).
The bold values highlight residues that can more likely be explored as possible transmission signatures that have a p-value < 0.05 and a q-value threshold < 0.3 to correct for multiple hypothesis testing, for which we used the Benjamini-Hochberg procedure to compute false discovery rate-adjusted p-values (q-values).
Association of Vif, Vpr, and Vpu mutations during early infection with known HLA epitopes
Given the reduced amino acid diversity of Vif, Vpr, and to a lesser degree Vpu in t/f viruses from our early infection cohort, we determined whether intrahost amino acid substitutions that arose in sequences collected up to 500 days p/s were associated with possible CTL responses in known HIV-1C-specific epitopes. 41 Four of the 17 patients exhibited amino acid substitutions within a previously described Vif CTL epitope restricted by their respective class I HLA during the follow-up period (Table 8). By 199 days p/s, a Vif-V98I substitution arose in patient B_2865 in a B*18:01 epitope. Furthermore, patient B_2865 harbored an E88Q substitution in the t/f virus, which persisted during the follow-up period. This substitution was not present in any of the 187 sequences analyzed from chronically infected patients, suggesting that B*18:01-mediated immune selection may have been responsible for the persistence of this variant. In patient E_3430, an A35T substitution within a known B*15:03 epitope arose in all Vif sequences by day 150 and persisted at least as far as day 372 p/s, at which K33E and F39S substitutions also predominated. Furthermore, the transmitted R34K and S36K variants, which we previously described as being more prevalent in HIV-1C t/f viruses, were maintained throughout the follow-up period in this individual, possibly due to HLA B*15:03 selection pressure.
CTL, cytotoxic T lymphocyte; HLA, human leukocyte antigen.
The bolded patient HLA represents the one associated with the CTL epitope.
Toward the end of the follow-up period, patient G_3603 developed E54A and K63I substitutions within a known A*68:01-restricted epitope, which occurred in cis as revealed by single-genome sequencing. Both of these amino acids were extremely rare in sequences from chronic infection, with 54A occurring in 1/187 sequences and 63I absent from the chronic dataset altogether. Patient QU_6029 also had a transmitted Vif variant, H56Y, which was extremely rare in chronic infection (1/187 sequences), yet persisted as the dominant variant within a B*15:10-restricted epitope at least as far as 174 days p/s. At that stage, a V51I variant emerged within the same B*15:10 epitope in 20% of the virus population.
Amino acid substitutions arising in early infection significantly reduce Vif and Vpr predicted epitope binding affinity
Due to the limited characterization of HLA-restricted CTL epitopes in HIV-1C accessory proteins, we used NetMHCpan 2.8 to predict autologous epitopes in Vif, Vpr, and Vpu and their binding affinities for each of the 17 patients. NetMHCpan, based on an artificial neural network, is reported to be the most accurate prediction currently available for MHC class I epitope binding. The output provides the predicted binding affinity of each putative epitope sequence to the restricting HLA allele. An IC50 value threshold of <500 nM was used to identify putative autologous epitopes.
We used the t/f virus from each patient to represent Vif, Vpr, and Vpu peptides in their putative immunologically susceptible (i.e., non-HLA adapted) form and each unique variant observed during the follow-up period as a putative escape (i.e., HLA-adapted) form. Considering each patient's HLA-A and HLA-B alleles and focusing on putative 9-mer epitopes, we evaluated any change in epitope binding affinity arising from a given amino acid substitution at each position within the epitope. We employed a sliding window approach whereby the query epitope was shifted by a single residue to avoid introducing bias by selecting only the epitopes with the strongest impact on HLA binding.
On average, amino acid substitutions seen in Vif during early infection increased the predicted median IC50 of putative epitopes from 151 [IQR 54–299] nM in the t/f viruses to 221 (IQR: 76–585) nM in the escaped form (p < .001, Wilcoxon matched-pairs signed rank test) (Fig. 4), suggesting that the escape form would exhibit lower affinity for the restricting HLA allele. All Vif substitutions that were predicted to alter MHC class I epitope binding affinity were plotted onto patient-specific maps; examples for two patients are illustrated in Figure 5, while maps for all other patients are included as Supplementary Figure S2. Overall, similar changes in predicted median IC50 of putative epitopes were observed for Vpr variants that arose during early infection (IC50 243 [IQR: 99–601 nM]) compared with those encoded in t/f viruses (192 [IQR 171–318] nM; p = .048) (Fig. 4). However, there was no significant difference in the binding affinity of putative Vpu epitopes found in the t/f viruses (214 [IQR: 94–564] nM) and those observed in early infection (240 [IQR: 84–452] nM; p = .65) (Fig. 4).
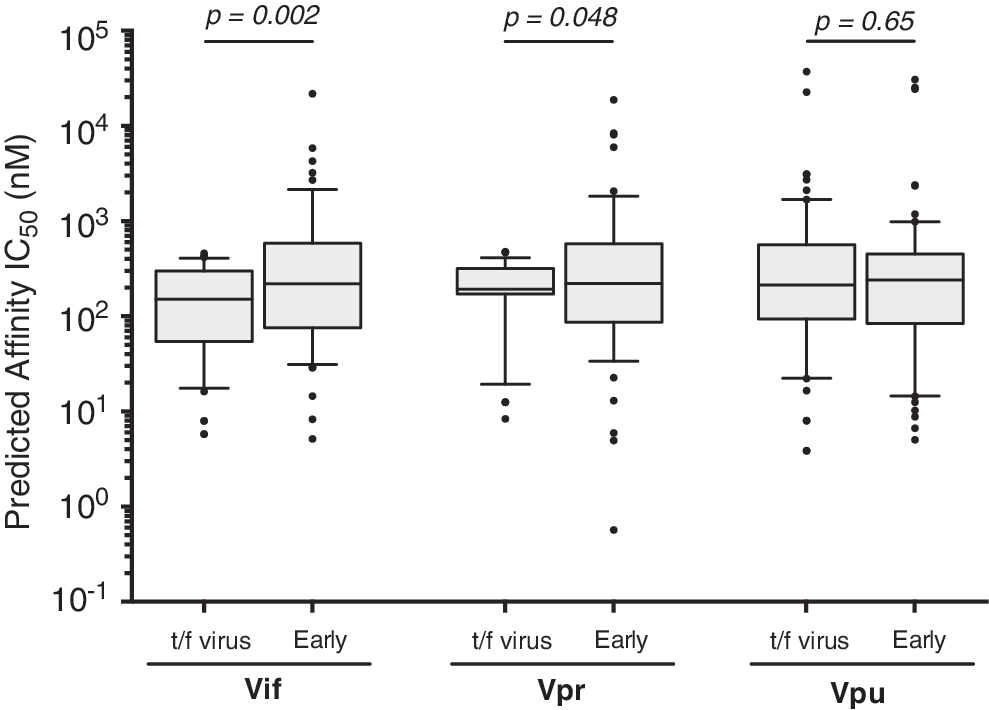
Predicted epitope binding affinity in putative HLA class I epitopes in t/f viruses and variants arising in early infection. NetMHCpan 2.8 was used to predict the binding affinities of all possible Vif, Vpr, and Vpu 9-mer epitopes for each of the 17 patients against their corresponding MHC class I HLA alleles. Affinities are given as the IC50 (nM), which is the peptide concentration at which half of the HLA molecules are occupied. As such, a lower IC50 for a peptide is indicative of a stronger binding affinity for the HLA in question. We used an IC50 cutoff of <500 nM to classify a putative peptide as being presented by HLA (Vif n = 58; Vpr n = 48; Vpu n = 69). Differences in binding affinity between putative epitopes in the t/f viruses were compared with the equivalent peptide found in early infection using the Wilcoxon matched-pairs signed rank test. HLA, human leukocyte antigen; MHC, major histocompatibility complex.

Predicted binding affinities of putative Vif epitopes during early infection. Two patient transmitted/founder (MRCA) and subsequent putative predicted epitope binding fold change illustrated for patients C and F. MRCA, most recent common ancestor.
We hypothesized that mutations were more likely to occur in anchor residues of 9-mer epitopes, defined by known HLA subtype-specific anchor motifs (
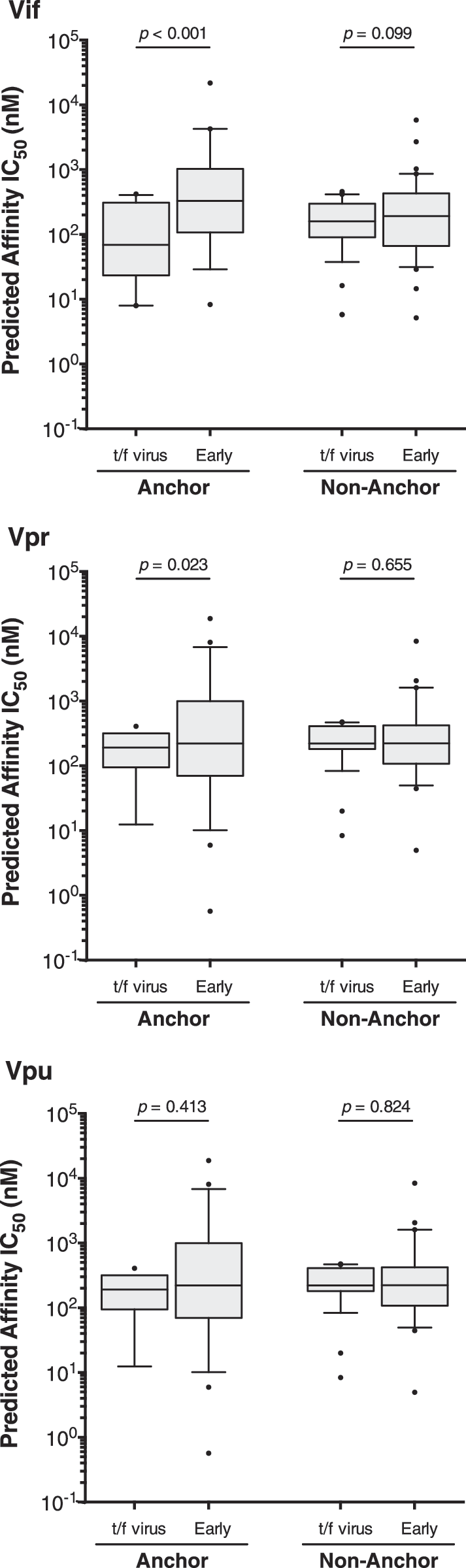
Difference in binding affinity for HLA class I epitope substitutions that occur in anchored compared with nonanchored position. Amino acid substitutions were characterized as occurring in anchor or nonanchor residues using HLA subtype-specific anchor motifs: Vif anchor n = 19, nonanchor n = 39; Vpr anchor n = 19, nonanchor n = 29; Vpu anchor n = 27, nonanchor n = 42. Predicated peptide binding affinities were compared using the Wilcoxon matched-pairs signed rank test.
Discussion
We assessed amino acid signature patterns in HIV-1C accessory genes over the first 500 days p/s and addressed the potential role of MHC class I HLA restriction in virus evolution during early HIV-1C infection. Unsurprisingly, most of the critical functional domains in Vif, Vpr, and Vpu were completely conserved in early HIV infection. Vif tryptophan residues 5, 11, 21, and 38, involved in APOBEC3G antagonism, were completely conserved among transmitted viruses, nor was there any variation in the 144-SLQYLA-149 motif, supporting Vif anti-APOBEC3G function as critical during early infection. 42 –49 Despite previous reports of low sequence variability, 36 Vif amino acid residues 63–70, required for the formation of β-strand structures and critical in maintaining normal vif expression levels, 50 showed slight variations in the t/f viruses to a similar degree as those published in the extended subtype C consensus 51 and the chronic infection subtype C analysis in this study. Residues Glu88 and Trp89 of the central hydrophilic region, which enhance steady-state expression of Vif, were conserved in the transmitted virus, although Gln88, previously uncharacterized in subtype C, was present in one patient instead of Glu88 (Val88 was the only alternative amino acid during chronic infection at 0.5%). A previous study, however, indicates that the hydrophilicity and charge of the central region are dispensable for the function of Vif 50 ; therefore, the presence of the weak basic amino acid Gln in place of acidic Glu may not affect steady-state expression. The 90-RLRR-93 motif was not conserved in the transmitted viruses similar to previously reported subtype C analyses. 36,52 The residues, Ser95 and Thr96, which are CKII and p44/42 mitogen-activated protein kinase phosphorylation sites, respectively, were relatively conserved in the transmitted viruses with variation present in one patient. As expected, the important His108, Cys114, Cys133, and His139 (HCCH) zinc-binding motif was completely conserved in the transmitted viruses from all 17 patients, as identified in previous subtype B and C analyses, 36,53,54 suggesting an important role for the HCCH-stabilized Vif–Cullin 5 interaction early in infection. The HIV-1 subtype B Vif phosphorylation site Thr155 is a highly conserved Lys155 in subtype C, which is 100% conserved in the transmitted viruses in this study, with variation observed in 2.7% of chronic infection sequences. Substitutions at this site could have an effect on phosphorylation and activity of Vif during HIV infection. The Vif oligomerization site 161-PPLP-164 was conserved throughout, confirming its central role in Vif function and viral infectivity during all stages of infection. At the phosphorylation site position 170, our findings are similar to previously published HIV-1 subtype C studies 36,52 where Val is the major amino acid in the transmitted viruses, although Ile is present in one patient.
Vpr t/f virus sequences were more conserved than those observed during chronic infection. The tertiary structure of Vpr comprises three α-helices folded around a hydrophobic core made up of leucine, isoleucine, valine, and aromatic residues. 55 The first α-helix (aa17-33) plays an essential role in nuclear localization and virion packaging, while the second (aa 38–50) is associated with oligomerization of Vpr and translocation of the preintegration complex to the perinuclear region. The third α-helix (aa 56–77) is associated with dimerization. Unlike previous findings in subtype C, 36,56 we found reduced conservation of Pro 10 and Pro 37 in the transmitted subtype C viruses. Try 54 and His71, previously described as aromatic amino acids involved in stabilizing the Vpr dimer and UNG2 binding, 36,56 were completely conserved in the t/f viruses. Mutations such as R77A/G, observed in the early HIV-1C infection cohort in this study, are known to reduce the proapoptotic activity of Vpr. 57,58 This variation is in line with the suggestion that proapoptotic activity of HIV-1 subtype C Vpr is different from HIV-1 subtype B. 36 R80A was not observed in any of 17 patients in this study and all transmitted viruses had arginine (R) at that position in Vpr. Therefore, it is plausible that in HIV-1 subtype C infection, two conserved arginine residues at positions 73 and 80 are sufficient for adenine nucleoside translocator interaction. 36 Other well-known substitutions, such as E21P, E24P, and A59P, which prevent the incorporation of Vpr into virus-like particles, as well as Q65R, which is correlated with impairment of Vpr docking at the nuclear envelope, 59 were not observed in this study. Neither were other known substitutions, underlying the integrity of Vpr during early HIV-1C infection. 60 –63
All 17 t/f viruses had a five-residue N-terminal insertion in Vpu compared with HIV-1 subtype B sequences. The transmembrane (TM) domain aa 12–29 and the hinge region residues 34–42 in the transmitted viruses were conserved, as previously described. 36 Differences in Gly38 and Ile41 positions were observed in the transmitted viruses in this study. Thr29 is predicted to act as a channel anchor and the presence of isoleucine in the transmitted viruses from five patients, a similar frequency as seen in chronic infection, could support the suggestion that the 100% conserved Tyr32 is an alternative channel anchor in subtype C. 36 The predicted regions thought to be associated with subtype C channel linking showed slight variations in Ile22 (one patient) and Ile23 (two patients), whereas Ile25 was 100% conserved as described previously. 36 Gly13 is thought to be involved in interacting with the membrane carbonyl oxygen groups to stabilize closed states and was present in the majority of transmitted viruses in our study, with alternative residue alanine present in three patients. Similar to findings by Bell et al., 36 no serine residues were observed in the TM domain, and the 7-RVDYR-11 was highly conserved, as previously described, excluding variation observed in Arg7, although the second-most prevalent amino acid at that position, Lys7, is also a highly charged positive amino acid, supporting the prediction that in HIV-1 subtype C, this is the region that maintains Vpu channel selectivity. 36 Vpu protein acts as an adaptor for CD4 proteasomal degradation by recruiting CD4 and β-transducin repeat-containing protein (βTrCP), the receptor component of the multisubunit SCF-βTrCP E3 ubiquitin ligase complex. The recognition signal for these cellular ligands by βTrCP is the phosphorylation of one or two serine residues present in a conserved Vpu motif, 57-DSGNES-62. 64 Vpu proteins in all transmitted viruses are predicted to bind βTrCP through Ser58 and Ser62, which were highly conserved in the transmitted Vpu. However, it is worth noting that overall, Vpu function is highly preserved despite sequence variation, which argues against the idea of a genetic signature for Vpu, which is suggested by our study. Additionally relevant, a recent study testing alleles from transmitted and chronic viruses by Mlcochova et al. 65 suggests that immune evasion activities of accessory proteins, Vpu and Vif, are conserved in acute and chronic HIV-1 infection. Note that at the q-value threshold of 0.3 used in the present study, it is expected that no >30% of identified associations would represent false positives; as such, further work is needed to confirm these putative transmitted versus chronic subtype C infection signatures.
Immune escape pathways are broadly predictable based on host HLA class I profile. 14 Nonconsensus variants (amino acid residues that differ from the HIV-1C consensus sequence) within known CTL-restricted epitopes in t/f virus sequences that were maintained over the first 500 days p/s cannot be conclusively associated with immune escape as these were presumably acquired at transmission. In contrast, variants that emerged within known or predicted CTL epitopes during follow-up are suggestive of immune escape 66 and are consistent with rapid initial accumulation of CTL escape mutations during the first weeks of infection. 18 The observation that many of these emerging variants are predicted to reduce peptide-HLA binding affinity further supports these as CTL escape mutations; 16,41,67 experimental validation of predicted epitopes and their selected variants will be addressed in future studies. It is also important to note that while we have used the terminology transmitted/founder viruses to describe the reconstructed MRCA HIV-1 sequence for each patient, we have not experimentally verified that these sequences are capable of being transmitted to multiple recipients, nor that the immunological pressures leading to changes in Vif, Vpr, and Vpu may have reduced the transmissibility of these viruses. Furthermore, to our knowledge, ours is the first study to investigate the impact of within-host evolution on the predicted binding affinities of putative novel epitopes in Vif, Vpr, and Vpu; as such, the generalizability of results to subtypes other than HIV-1C remains to be determined.
Our study was further constrained by the limited characterization of CTL epitopes in HIV-1 subtype C, particularly in accessory proteins. To partially address this limitation, we employed an artificial neural network-based prediction of epitope:HLA binding to identify putative epitopes. By comparing the affinity of predicted epitopes within the transmitted Vif, Vpr, and Vpu sequences to variants that arose within the patient quasispecies over 500 days p/s, we determined that polymorphisms in Vif and Vpr were more likely to significantly decrease epitope binding when the mutation occurred in an anchor residue. These results highlight the need for further quantitative work on the impact of CTL escape mutations in HIV-1C accessory proteins.
In summary, our results highlight (1) highly conserved regions in accessory genes during both primary and chronic HIV-1 subtype C infection, (2) putative transmission signature residues in Vif and Vpr, but not Vpu, (3) the suggestion of possible escape mutations that are HLA-associated early p/s, and (4) that primary infection polymorphisms in Vif and Vpr accessory genes were more likely to decrease epitope binding when the mutation occurred at an anchor residue. These data contribute to the knowledge about primary HIV-1 subtype C infection and could be helpful toward HIV vaccine strategies that incorporate accessory genes as components.
Footnotes
Acknowledgments
The authors are grateful to the participants of the Tshedimoso study in Botswana—without their participation, this research would not have been possible (Re a leboga le kamoso). The authors thank the Botswana Ministry of Health, Gaborone City Council clinics, and the Gaborone VCT Tebelopele for collaboration and are grateful to Gaseboloke Mothowaeng, Florence Modise, S'khatele Molefhabangwe, Sarah Masole, and the late Melissa Ketunuti for their dedication and outstanding work in the clinic and outreach. The authors are grateful to Jan Walter De Neve, Lauren Margolin, Lauren Buck, Andrew J. Fiore-Gartland, Jeannie Baca, and Brian Foley for excellent technical assistance and thank Lendsey Melton for editorial assistance. They thank the Botswana Harvard AIDS Institute (Gaborone, Botswana) and the Essex Lab (Harvard T.H. Chan School of Public Health, Boston, MA) where the laboratory and data analyses were performed. The primary HIV-1 subtype C infection study in Botswana, the Tshedimoso study, was supported by the National Institute of Allergy and Infectious Diseases (R01 AI057027). R.R. was supported, in part, by the NIH Fogarty International Clinical Research Scholars and Fellows Program, the NIH Fogarty International Center AIDS International Training and Research Program (D43 TW000004), and the NIH Fogarty HIV Research Training Program for Low- and Middle-Income Countries (D43 TW009610). I.J.M. was supported by a Feasibility Award from the Harvard University Center for AIDS Research (CFAR), an NIH-funded program (P30 AI060354). Z.L.B. was supported by a New Investigator Award from the Canadian Institutes of Health Research (CIHR) and a Scholar Award from the Michael Smith Foundation for Health Research (MSFHR). This work was also supported, in part, by University of Botswana ORD, R812 (TKS).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.