
Abstract
We report that the addition of an host paracaspase MALT1 inhibitor, MI-2, to HIV latently infected ACH-2, Jurkat E4, and J-LAT cells accelerated cell death in the presence of cell stimuli or the protein kinase C agonist, bryostatin 1. MI-2-mediated cell death correlated with the induction of the cellular RNase MCPIP1 and requires the presence of viral component(s). Altogether, the combination of MI-2 and bryostatin 1 displays selective killing of HIV latently infected CD4+ T cells.
H
We have previously reported that a cellular RNase monocyte chemotactic protein-induced protein 1 (MCPIP1) restricts HIV-1 infection in resting CD4+ T cells. 10 Interestingly, MCPIP1 is rapidly degraded in activated primary T cells. 10 We 11 and others 12 subsequently demonstrated that MCPIP1 was cleaved in activated human and mouse CD4+ T cells by the mucosa-associated lymphoid-tissue lymphoma-translocation gene 1 (MALT1), a paracaspase whose activity is critically important for activation of T and B lymphocytes. 13,14 MALT1 cleaves MCPIP1 at the C-terminal side of an arginine residue of the PEST sequence found in its substrates, including Bcl10, CYLD, and A20. 15 Of note, MCPIP1 knockout mice displayed hyperactivation of CD4+ T cells, including memory CD4+ T cells. 12,16
Based on these findings, we postulated that blocking MALT1-dependant MCPIP1 cleavage in activated CD4+ T cells may restore MCPIP1 levels and confer resistance to HIV-1. Among several reported MALT1 inhibitors, MI-2 was shown to selectively bind to and inhibit the cleavage activity of MATL1. 17 MI-2 contains a reactive chloromethyl amide and covalently binds to and irreversibly blocks MALT1 cleavage activity (Fig. 1A, B). 17 To examine the effect of MI-2 on MALT1-mediated MCPIP1 cleavage, we treated Jurkat T cells with MI-2 and found that MCPIP1 is rapidly upregulated on addition of MI-2 (Fig. 1C). Interestingly, the protein levels of another two MALT1 substrates, A20 and CYLD, either modestly changed or did not change at all following MI-2 treatment.
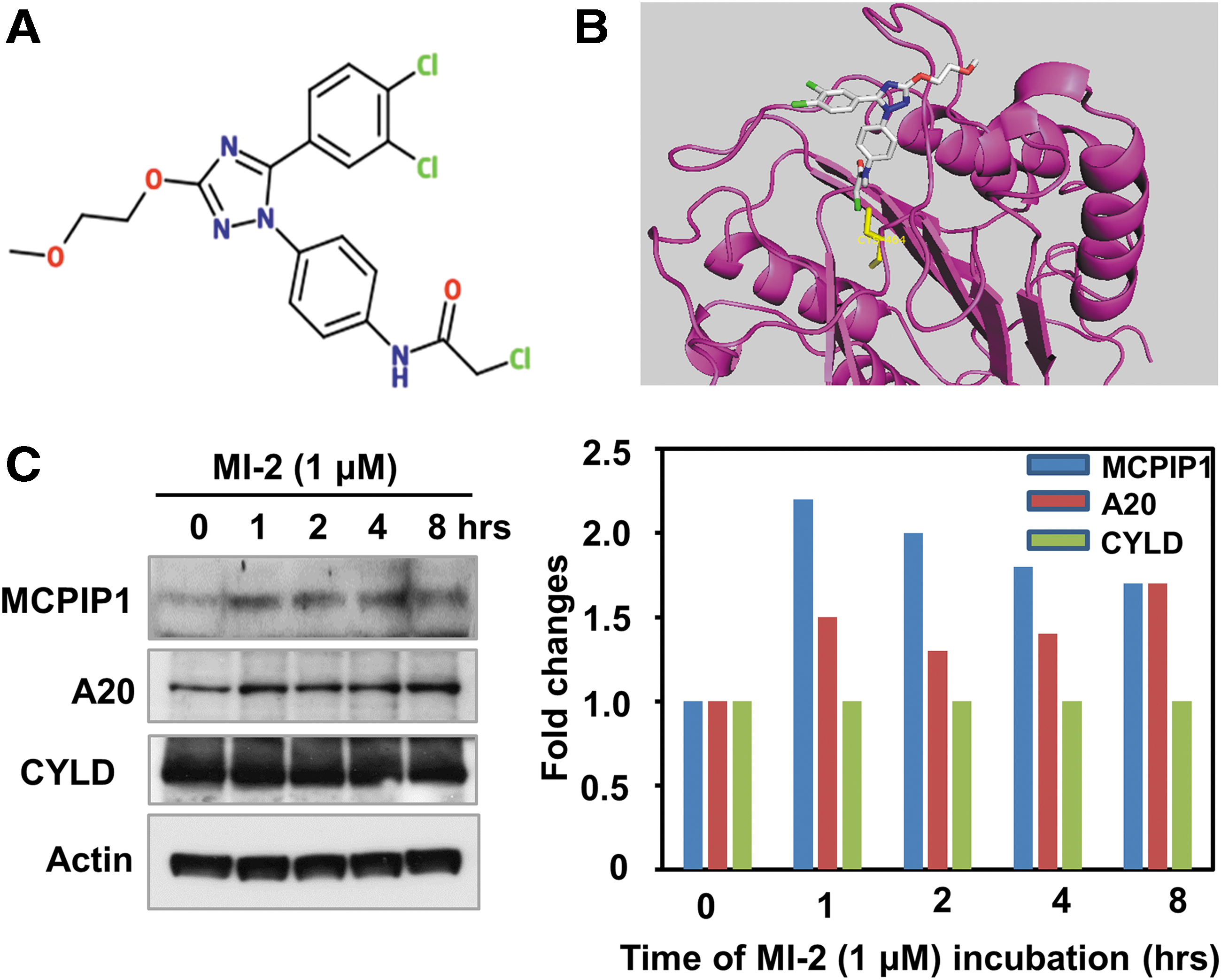
MI-2 induces MCPIP1 expression in Jurkat T cells.
Next, we sought to test whether MI-2 treatment confers resistance to HIV in reactivated HIV latently infected cells. We chose to work with three cell line-based HIV latency models, including the J-Lat Tat-GFP (A2 clone) and J-Lat full length 9.2 clone from Verdin's laboratory,
18
the Jurkat E4 clone from Karn's group,
19
and the ACH-2 clone,
20
which all harbor a latent HIV provirus in various forms. To our surprise, although MI-2 is nontoxic to animals, it induced massive cell death in cell line-based HIV latency models when cell activation signals were supplied (Supplementary Fig. S1; Supplementary Data are available online at
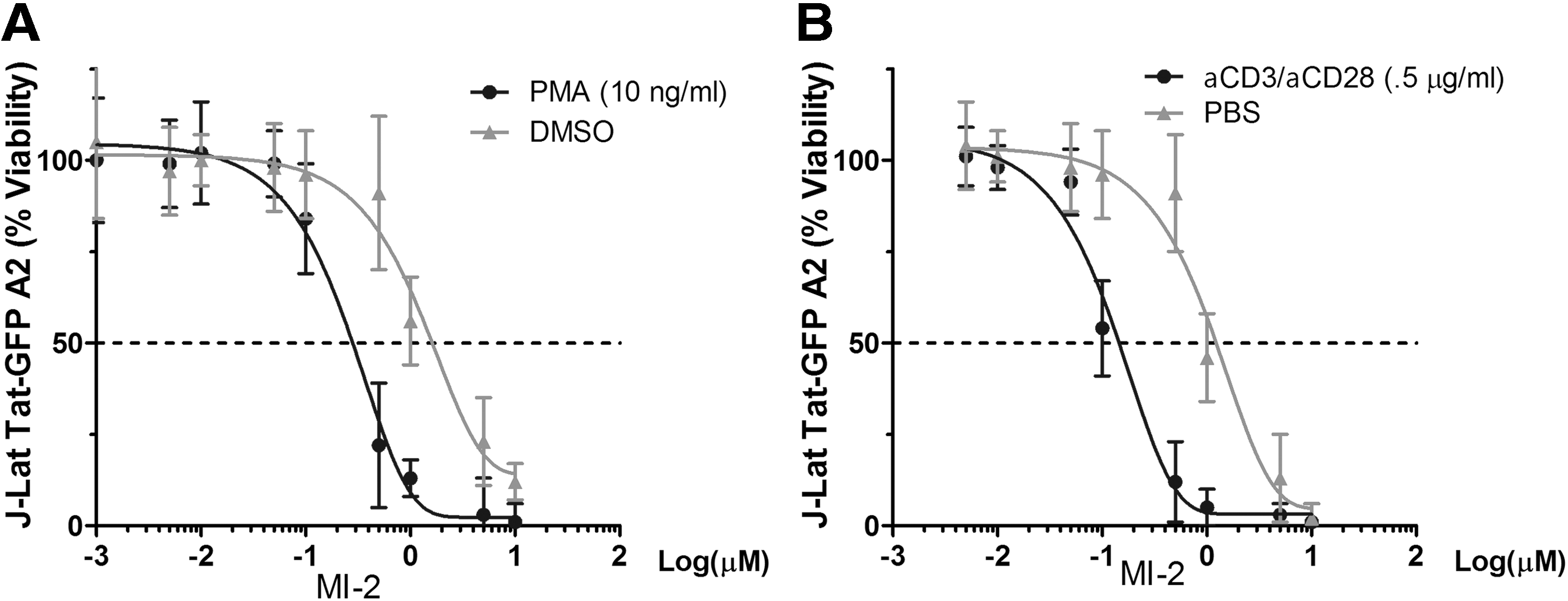
Cell activation increases MI-2-mediated cell death in HIV latently infected cells.
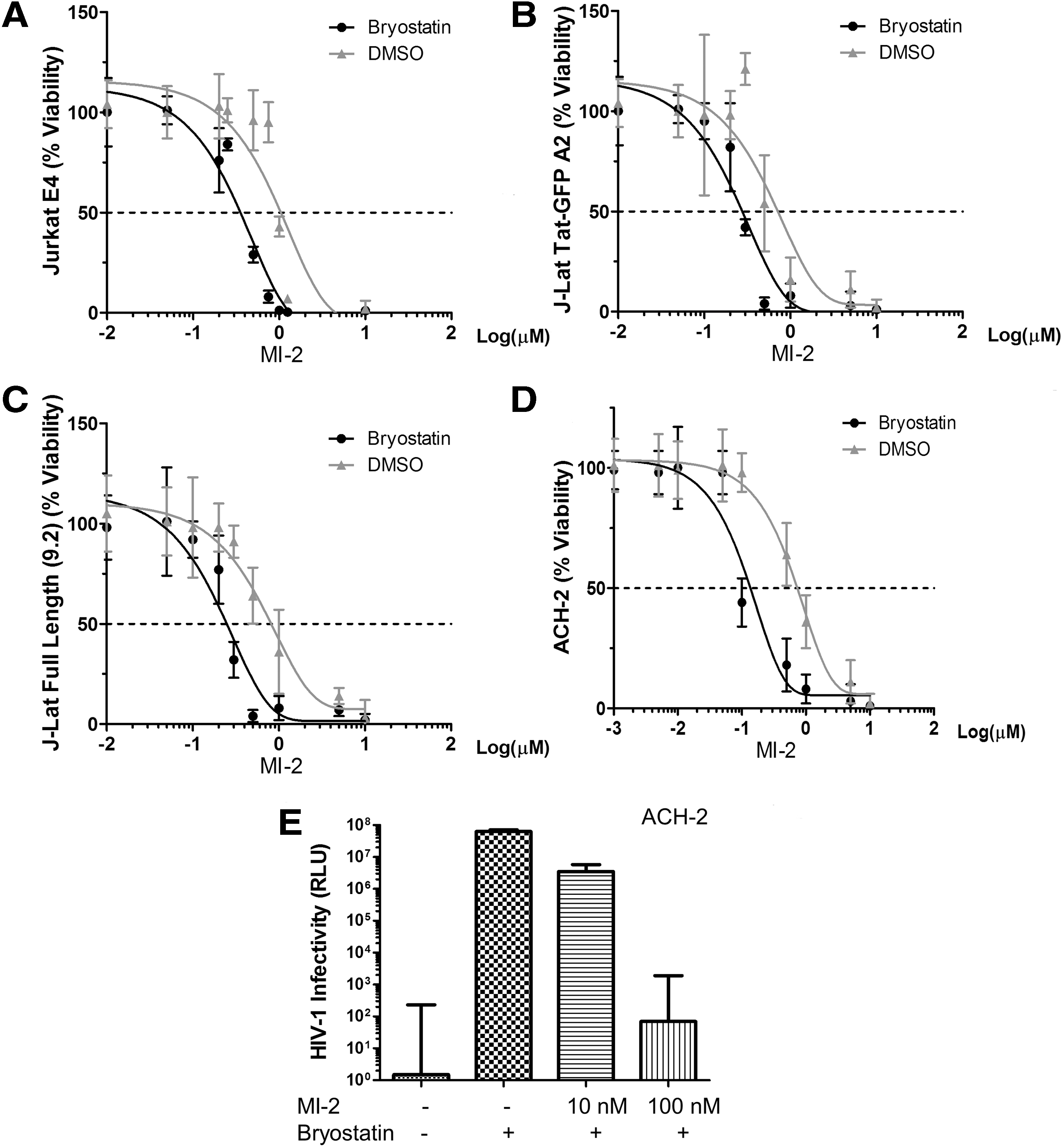
Bryostatin 1 increases MI-2-mediated cell death in HIV latently infected cells.
Finally, we determined whether MI-2 in combination with bryostatin 1 selectively targets reservoir with lethal effect. As shown in Figure 4A and B, MI-2 in combination with bryostatin 1 does not accelerate cell death of uninfected Jurkat T nor CEM cells, suggesting that the accelerated cell death achieved in the HIV latently infected cell lines involves viral components. The question is whether the observed cell death is exclusively dependent on MCPIP1 induction. Previously we have shown that elevated MCPIP1 enhanced stress-induced apoptosis in RAW264.7 cells. 22 We speculate that HIV latently infected CD4+ T cells are more prone to MCPIP1-mediated cell death on reactivation. Since MCPIP1 is an RNase and shown by others to directly bind viral RNA, 23 newly transcribed HIV mRNA species in the reactivated cells may trigger MCPIP1-mediated cell death. In other words, elevated MCPIP1 on MI-2 treatment detects HIV mRNA species and causes cell death. If true, such a mechanism may only require the transcriptional reactivation of proviruses, which can be easily achieved by LRAs. Ongoing experiments are underway to test this possibility.
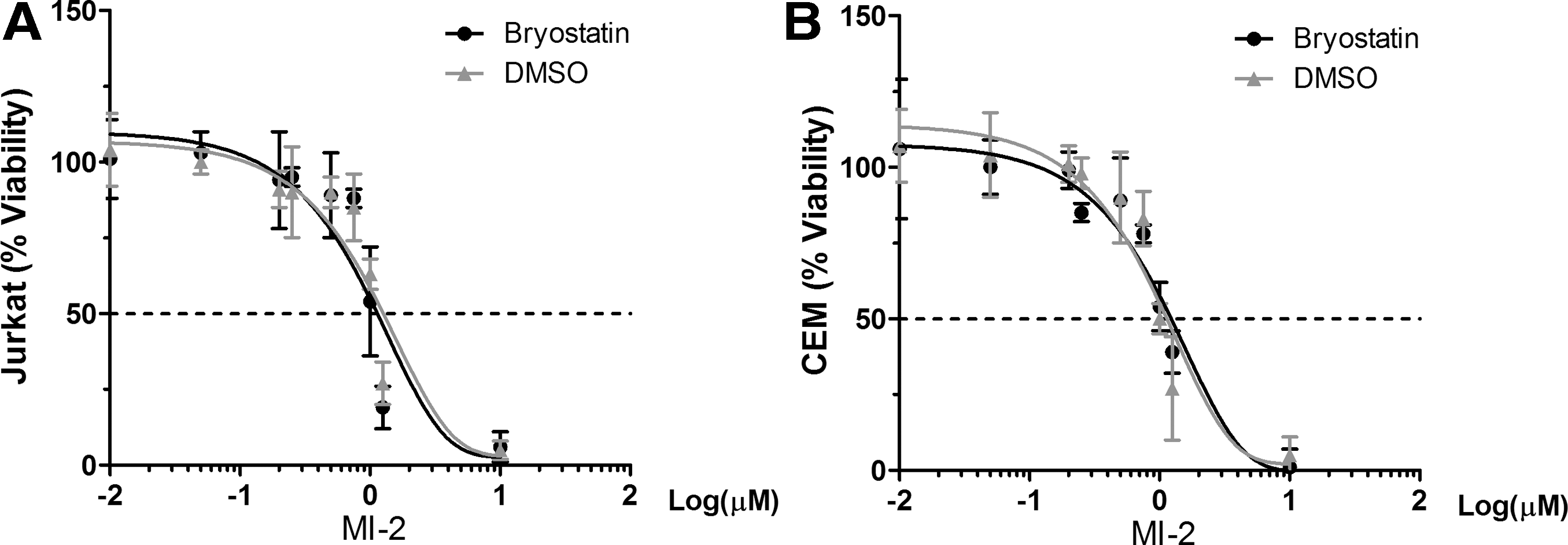
Bryostatin 1 does not increase MI-2-mediated cell death in uninfected Jurkat T
Collectively, our data suggest that the MALT1 inhibitor in combination with LRA represents a novel approach to kill HIV latently infected T cells. The therapeutic index (TI, ∼10) of MI-2 warrants in-depth follow-up analysis. Interestingly, two studies of MALT1 inhibitors revealed that MALT1 cleavage activities can also be achieved through reversible binding of MALT1 by one MI-2 analog, MI-2A3, 17 and another small molecule named mepazine. 24 These compounds may offer wider therapeutic window because reversible inhibition of MALT1 is expected to cause less death to bystander cells. Further investigations of these compounds using primary latency model and ex vivo HIV-1 latency model will confirm the validity of this novel approach.
Footnotes
Acknowledgments
This study was sponsored by the National Institute of Health Grant R01DK088787 and R56DK088787 (to T.T.W.) and by the Natural Science Foundation of Heilongjiang Province grant QC2012C094 (to H.L.). M.F was supported by the National Institute of Health Grant R21AI103618. H.L. is a recipient of the “Reserve Talents of Universities Overseas Research Program of Heilongjiang Education Department.” The funders had no role in the study design, data collection, and interpretation, or the decision to submit the work for publication. The authors wish to thank Dr. Fatah Kashanchi for providing reagents and helpful advice. The J-Lat and ACH-2 clones were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: J-Lat Full Length GFP Cells from Dr. Eric Verdin and Dr. Thomas Folks.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.