
Abstract
Cessation of highly active antiretroviral therapy (HAART) in HIV-infected individual leads to a rebound of viral replication due to reactivation of a viral reservoir composed largely of latently infected memory CD4+ T cells. Efforts to deplete this reservoir have focused on reactivation of transcriptionally silent latent proviruses. HIV provirus transcription depends critically on the positive transcription elongation factor b (P-TEFb), whose core components are cyclin-dependent kinase 9 (CDK9) and cyclin T1. In resting CD4+ cells, the functional levels of P-TEFb are extremely low. Cellular activation upregulates cyclin T1 protein levels and CDK9 T-loop (T186) phosphorylation. The broad-spectrum histone deacetylase inhibitors (HDACis) vorinostat and panobinostat have been shown to reactivate latent virus in vivo in HAART-treated individuals. In this study, we have found that vorinostat and panobinostat activate P-TEFb in resting primary CD4+ T cells through induction of CDK9 T-loop phosphorylation. In contrast, tacedinaline and romidepsin, HDAC 1 and 2 inhibitors, were unable to activate CDK9 T-loop phosphorylation. We used a CCL19 primary CD4+ T-cell model HIV latency to assess the correlation between induction of CDK9 T-loop phosphorylation and reactivation of latent HIV virus by HDACis. Vorinostat and panobinostat treatment of cells harboring latent HIV increased CDK9 T-loop phosphorylation and reactivation of latent virus, whereas tacedinaline and romidepsin failed to induce T-loop phosphorylation or reactivate latent virus. We conclude that the ability of vorinostat and panobinostat to induce latent HIV is, in part, likely due to the ability of the broad-spectrum HDACis to upregulate P-TEFb through increased CDK9 T-loop phosphorylation.
P
Compounds of diverse chemical nature such as the differentiation agent HMBA, histone deacetylase inhibitors (HDACis), bromodomain and extraterminal bromodomain inhibitors, and protein kinase c (PKC) agonists can activate HIV-1 transcription from either latent cell lines or from resting primary CD4+ T cells. 13 –17 Treatment of resting CD4+ T cells with 335 nM of the broad-spectrum HDACi, vorinostat, which is equivalent to a single in vivo dose of 400 mg, activated latent HIV-1 in patients' cells ex vivo. 18 Vorinostat was also evaluated for its ability to reactive the latent HIV-1 in vivo and it was shown to reactivate latent HIV-1 in vivo in patients in two studies. 18,19 Despite activation of latent virus by vorinostat, no reduction in the size of the latent HIV reservoir was observed in patients in either of these two studies. In addition, the broad-spectrum HDACi panobinostat has been shown to reactivate latent HIV in patients in vivo, but like vorinostat, panobinostat failed to reduce the level of the latent HIV reservoir. 20
Recent data from our laboratory demonstrated that vorinostat activates P-TEFb in primary resting CD4+ T cells through an increase in CDK9 T-loop phosphorylation. 21 In this present study, we examined whether other HDACis can also activate P-TEFb and if there exists a relationship between P-TEFb activation and increased histone H3 acetylation. To determine whether vorinostat and the additional broad-spectrum HDACi panobinostat can activate P-TEFb through an increased phosphorylation of CDK9 and whether this effect is dose dependent, resting CD4+ T cells were incubated with increasing concentrations of different HDACis for 24 h, cell extracts were prepared as previously described, 8 and CDK9 was analyzed in immunoblots (Fig. 1). Vorinostat and panobinostat treatment of CD4+ T cells increased the CDK9 T-loop phosphorylation in a dose-dependent manner. We did not observe a reproducible increase in the basal expression level of cyclin T1 by either vorinostat or panobinostat (data not shown). Vorinostat and panobinostat treatment also increased the acetylation histone H3 in a dose-dependent manner compared to untreated cells. In contrast, treatment of CD4+ T cells with either tacedinaline or romidepsin, both of which are HDAC 1 and 2 inhibitors, failed to increase of CDK9 T-loop phosphorylation or increase cyclin T1 levels (Fig. 1). Although romidepsin failed to increase the phosphorylation of CDK9, we found a dose-dependent increase of histone H3 acetylation in CD4+ T cells. Only a very modest increase in histone H3 acetylation was observed for tacedinaline; there are reports that tacedinaline can cause hyperacetylation of histone H3 in HCT-8 colon carcinoma cells, but histone H3 acetylation occurred only at high concentrations (750 nM) in the previous study 22 ; the IC50 of tacedinaline is 0.57 μM for HDAC1. Thus, our data show that the broad-spectrum HDACis, vorinostat and panobinostat, induced CDK9 T-loop phosphorylation in resting CD4+ T cells, while the HDAC1 and HDAC2 inhibitors, tacedinaline and romidepsin, did not.
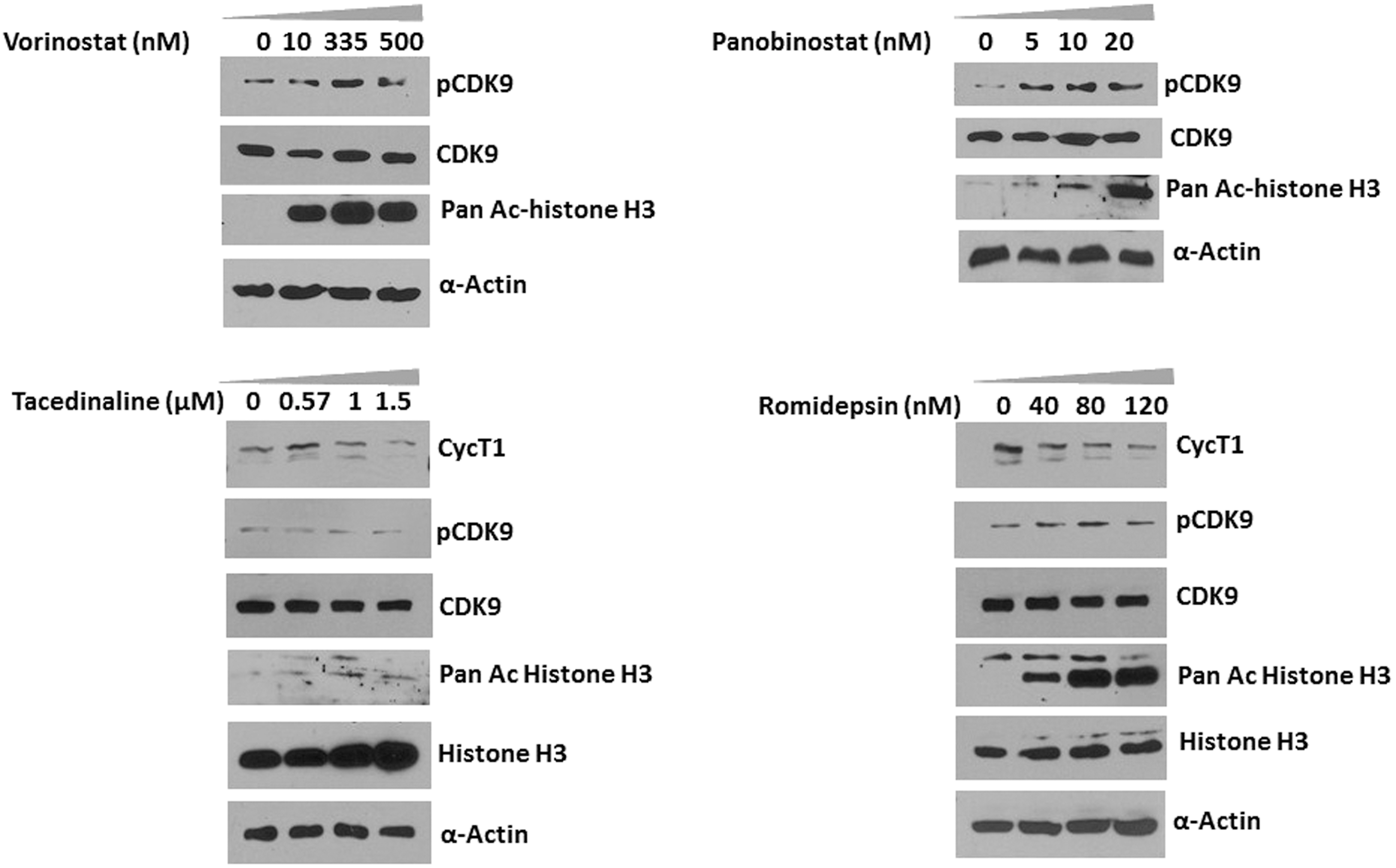
Vorinostat (SAHA) and panobinostat induce CDK9 Thr-186 T-loop phosphorylation in resting CD4+ T cells. Resting CD4+ T cells were isolated by negative selection from peripheral blood of healthy donors by the Rosettesep CD4+ cells isolation kit (STEMCELL Technologies, Inc.). Flow cytometry analysis confirmed that the CD4+ cell populations were routinely >98% (data not shown). Cells were treated with various concentrations of vorinostat, panobinostat, tacedinaline, or romidepsin for 24 h and lysates were prepared in EBCD buffer [50 mM Tris-HCl (pH 8.0), 120 mM NaCl, 0.5% Nonidet P-40, 5 mM dithiothreitol, 4 mM MgCl2 buffer containing protease inhibitor cocktail] and analyzed for the expression of indicated proteins in immunoblots. Experiments were repeated at least three times and representative figures are shown. CDK9, cyclin-dependent kinase 9.
Next, we examined the time course of CDK9 T-loop phosphorylation in resting CD4+ T cells by either vorinostat (335 nM) or panobinostat (10 nM). Cell lysates were prepared at different time points of HDACi treatment, and lysates were examined in immunoblots. As shown in Figure 2, increased CDK9 Thr-186 phosphorylation by both vorinostat and panobinostat was first observed at 12 h post-treatment. CDK9 T-loop phosphorylation increased at 18 h and remained high at 24 h of HDACi treatment. Interestingly, increased acetylation of histone H3 was observed at 3 h by panobinostat but not vorinostat; increased H3 acetylation by vorinostat was first observed at 12 h post-treatment, similar to the kinetics of CDK9 T-loop phosphorylation by vorinostat. Thus, vorinostat and panobinostat induce significant levels of CDK9 T-loop in resting CD4+ T cells at 12 h post-treatment. The kinetics of increased T-loop phosphorylation and histone H3 acetylation is similar for vorinostat, whereas for panobinostat, histone H3 acetylation is more rapid than T-loop phosphorylation.
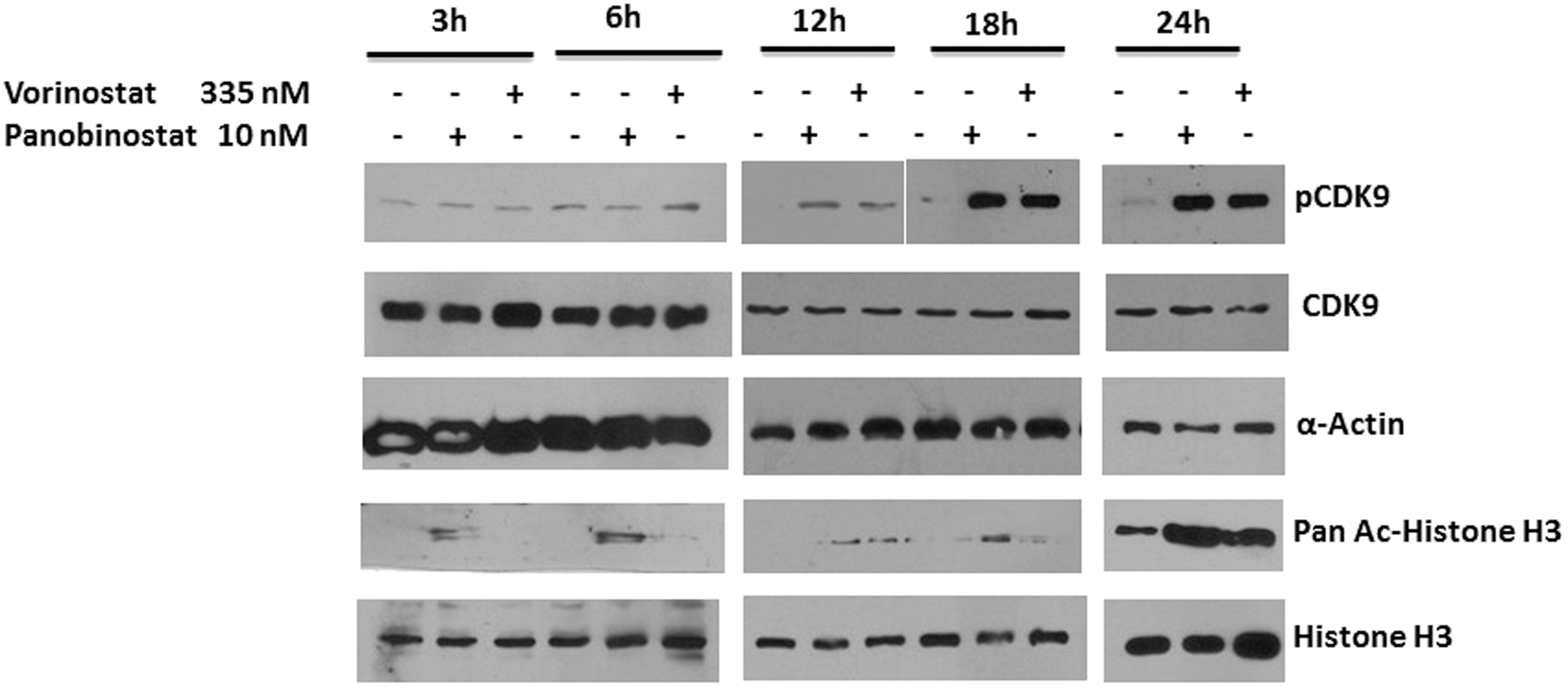
Time course of CDK9 T-loop phosphorylation in HDACi-treated CD4+ T cells. CD4+ T cells prepared as described in Figure 1 were treated with either vorinostat or panobinostat for the indicated time periods. Cell lysates were prepared at the indicated times as described in Figure 1 and lysates were examined in immunoblots. HDACi, histone deacetylase inhibitor.
We next determined whether the induction of CDK9 T-loop phosphorylation by vorinostat and panobinostat might correlate with reactivation of latent HIV-1 in a primary CD4+ T-cell model. We therefore used the CCL19 chemokine latency model. 23,24 In this model, resting CD4+ T cells are treated with CCL19, which establishes conditions in which the cells support high levels of latent infection that can be subsequently reactivated by T-cell activation. We generated stocks of vesicular stomatitis virus (VSV) pseudotyped HIV-1 reporter virus, which contain the Luciferase coding sequences in place of the nef gene and inactivating mutations in the env and vpr genes (obtained from the NIH AIDS Reagent program). Resting CD4+ T cells were treated with the chemokine CCL19 for 72 h followed by infection with the reporter virus for another 5 days. As shown in Figure 3A and 3B, CCL19 treatment alone did not induce T-loop phosphorylation or cyclin T1 levels. In contrast, phorbol 12-myristate 13-acetate (PMA) plus ionomycin treatment increased T-loop phosphorylation and the level of cyclin T1. Both vorinostat and panobinostat, but not tacedinaline, increased CDK9 T-loop phosphorylation in CCL19-treated cells (Fig. 3B) compared to control cells. Vorinostat and panobinostat, but not tacedinaline, also increased histone H3 acetylation in CCl19-treated cells. In agreement with the induction of CDK9 T-loop phosphorylation, both vorinostat treatment and panobinostat treatment, but not tacedinaline, were able to significantly reactivate latent virus as determined by induction of Luciferase expression (Fig. 3C). In an additional experiment shown in Figure 3D, tacedinaline and romidepsin failed to demonstrate a significant reactivation of latent HIV, while vorinostat and panobinostat demonstrated a significant reactivation of latent HIV similar to that observed in the experiment shown in Figure 3C. We note that tacedinaline treatment resulted in an increase in the minor 55 kDa form of CDK9 in the experiment shown in Figure 3D (band above majo 42 kDa form of CDK9). 25,26 The significance of this increase is unclear. We conclude from these data that vorinostat and panobinostat can reactivate latent virus in the CCL19 model of latency and this correlates with an induction of CDK9 T-loop phosphorylation.
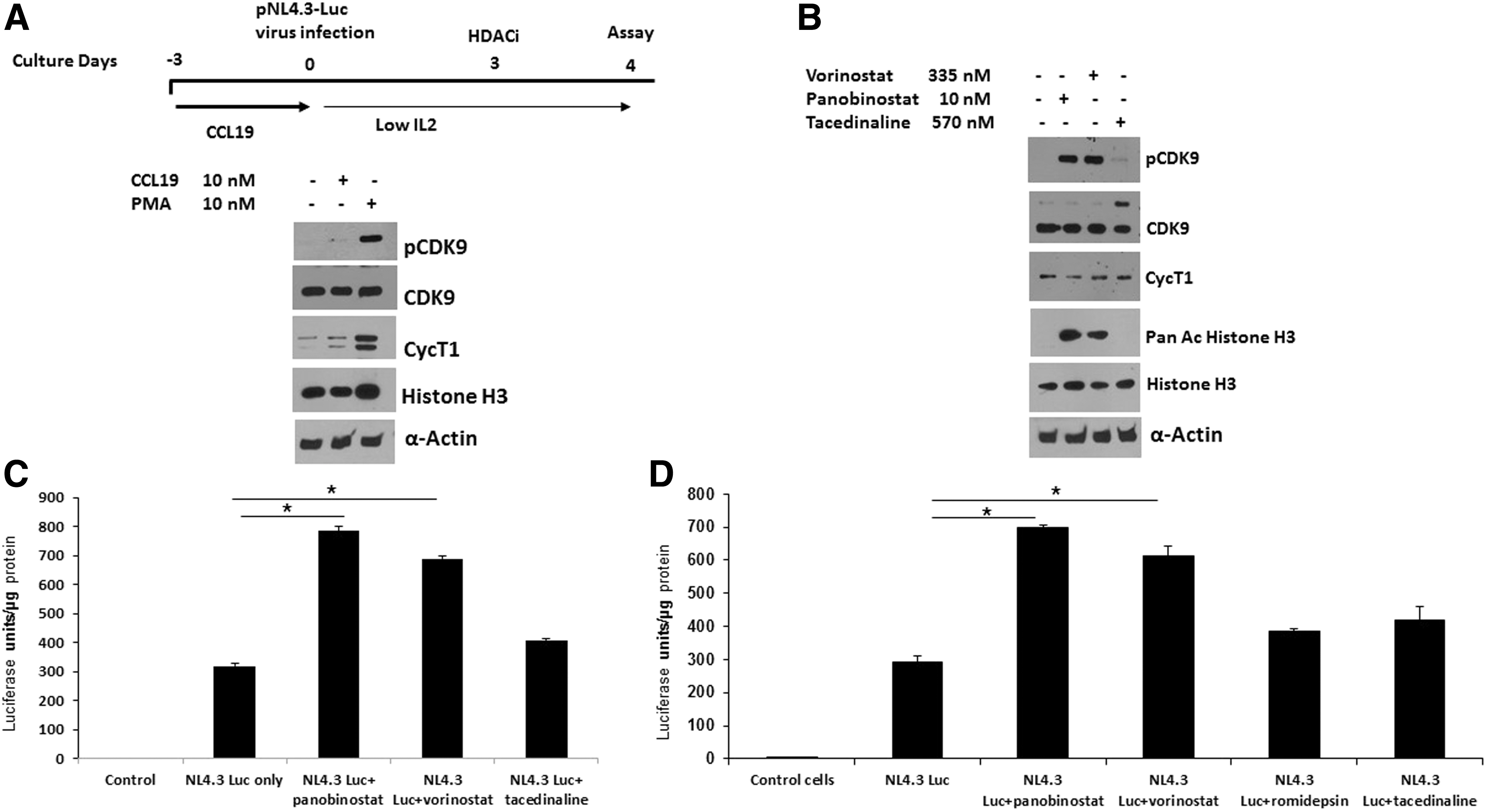
Vorinostat and panobinostat activate HIV-1 in CCL19-mediated HIV-1 latency model in resting CD4+ T cells.
In summary, this study demonstrates that the broad-spectrum HDAC inhibitors vorinostat and panobinostat can reactivate latent HIV-1 to some extent and this is likely due to counteracting repressive chromatin through increased histone acetylation as well as an induction of P-TEFb activity through an increase in CDK9 T-loop phosphorylation. However, the data to date from the field indicate that single agents such as vorinostat or panobinostat will not be effective when used alone in reactivating latent HIV in vivo. 27 An effective reactivation strategy will require multiple agents that act through distinct mechanisms to relieve a repressive chromatin environment for latent viruses and induce limiting levels of cellular transcription factors, especially P-TEFb. In addition, reduction of the reservoir will require strategies to enhance the immune system's ability to recognize and clear cells in which latent viruses have been reactivated. 28 Further studies on the mechanism through which vorinostat and panobinostat induced CDK9 T-loop phosphorylation may provide clues for potent latency reactivation strategies.
Footnotes
Acknowledgment
This work was supported by the National Institutes of Health grants AI110263 and AI116173 (to APR) and P30AI1036211 (Baylor-UT-Houston CFAR).
Author Disclosure Statement
No competing financial interests exist.