
Abstract
Human immunodeficiency virus type 1 (HIV-1) escapes complement-mediated lysis (CML) by incorporating host regulators of complement activation (RCA) into its envelope. CD59, a key member of RCA, is incorporated into HIV-1 virions at levels that protect against CML. Since CD59 is a glycosylphosphatidylinositol-anchored protein (GPI-AP), we used GPI anchor–deficient Jurkat cells (Jurkat-7) that express intracellular CD59, but not surface CD59, to study the molecular mechanisms underlying CD59 incorporation into HIV-1 virions and the role of host proteins in virus replication. Compared to Jurkat cells, Jurkat-7 cells were less supportive to HIV-1 replication and more sensitive to CML. Jurkat-7 cells exhibited similar capacities of HIV-1 binding and entry to Jurkat cells, but were less supportive to viral RNA and DNA biosynthesis as infected Jurkat-7 cells produced reduced amounts of HIV-1 RNA and DNA. HIV-1 virions produced from Jurkat-7 cells were CD59 negative, suggesting that viral particles acquire CD59, and probably other host proteins, from the cell membrane rather than intracellular compartments. As a result, CD59-negative virions were sensitive to CML. Strikingly, these virions exhibited reduced activity of virus binding and were less infectious, implicating that GPI-APs may be also important in ensuring the integrity of HIV-1 particles. Transient expression of the PIG-A gene restored CD59 expression on the surface of Jurkat-7 cells. After HIV-1 infection, the restored CD59 was colocalized with viral envelope glycoprotein gp120/gp41 within lipid rafts, which is identical to that on infected Jurkat cells. Thus, HIV-1 virions acquire RCA from the cell surface, likely lipid rafts, to escape CML and ensure viral infectivity.
Introduction
N
It is believed that HIV-1 acquires biologically functional RCA proteins during viral budding from the plasma membrane of infected cells. Incorporation of these host RCA proteins into HIV-1 virions is not random or simply a function of expression level or density on the cell surface since proteins that are highly expressed on infected cells, such as CD4, CD45, CXCR4, CCR3, and CCR5, are not incorporated into HIV-1 virions. 3,30 –32 However, the precise mechanism(s) by which RCA proteins are selectively incorporated onto HIV-1 particles is not clearly understood. CD46 is a single-pass type I membrane protein that has its N-terminus exposed to the extracellular or luminal space, while CD55 and CD59 are two glycosylphosphatidylinositol-anchored proteins (GPI-APs). Compared to CD46, CD55 and CD59 are more effective in suppressing ADCML of HIV-1 virions. Blockage of CD55 and CD59 with their specific Abs strongly enhances ADCML of HIV-1 virions, whereas blockage of CD46 has a significantly weaker effect. 10 As members of the GPI-AP family, CD55 and CD59 proteins lack a transmembrane domain, have no cytoplasmic tail, and are, therefore, located on the outer leaflet of the plasma membrane. 33
The GPI anchor precursors are synthesized in the ER, and at least 26 gene products are involved in the process. 34 Among these genes, the PIG-A gene is essential for the biosynthesis of GPI-APs. In humans, somatic mutations in the coding region of the X-linked PIG-A gene can occur in early hematopoietic stem cells. Nonmalignant clonal expansion of hematopoietic stem cells with somatic mutations in the PIG-A gene causes paroxysmal nocturnal hemoglobinuria (PNH), a rare form of acquired hematologic disorder characterized clinically by intravascular hemolysis and venous thrombosis. 35,36 The PIG-A gene encodes an enzyme phosphatidylinositol N-acetylglucosaminyltransferase subunit A, which is required for the synthesis of N-acetylglucosaminyl-phosphatidylinositol (GlcNAc-PI), the first intermediate in the biosynthetic pathway of the GPI anchor. As PIG-A participates in the early steps of GPI anchor biosynthesis, a defect or mutation in the PIG-A gene results in deficiency of GPI anchors and subsequent impairment of the surface expression of CD55 and CD59, hence predisposing the cells sensitive to complement attack.
This study was undertaken using GPI anchor–deficient Jurkat cells (Jurkat-7), which lack CD59 on the cell surface, as a model to study the role of GPI-APs, particularly CD59, in the HIV-1 life cycle, incorporation of host proteins into virions, and complement-mediated lysis (CML) of virions. Our results indicate that HIV-1 virions incorporate CD59 from the host cell surface rather than the intracellular compartments. Deficiency of GPI-APs on the cell surface attenuates the production of infectious HIV-1 virions and contributes to virions that are more susceptible to CML. Since GPI-APs and gp120/gp41 are colocalized in lipid rafts, HIV-1 virions likely acquire RCA from the lipid rafts on the cell surface to escape CML and ensure viral infectivity.
Materials and Methods
Cell lines and HIV-1 stocks
Jurkat cells (P2 strain), a human leukemia T-lymphocyte cell line, were purchased from ATCC (Manassas, VA). Jurkat-7 cells, a GPI anchor–deficient mutant of Jurkat cells, were provided by Dr. J. Schubert (Hannover Medical School, Hannover, Germany) as a generous gift.
37
The TZM-bl cell line, a HeLa cell derivative, was obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD). TZM-bl cells were genetically engineered to express CD4, CCR5, and CXCR4
38
and to contain Tat-responsive reporter genes for firefly luciferase (Luc) and Escherichia coli β-galactosidase under the control of an HIV-1 long terminal repeat.
39
These engineered features made TZM-bl cells highly susceptible to infection by most laboratory strains and primary isolates of HIV-1.
40
Jurkat, Jurkat-7, and TZM-bl cell lines were cultured in the complete RPMI 1640 medium containing 10% fetal bovine serum (FBS; Atlanta Biological, Atlanta, GA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM
Complement-mediated cytolysis and virolysis
To analyze complement-mediated cytolysis, approximately 0.5 × 106 Jurkat or Jurkat-7 cells were exposed to normal human serum (NHS, complement competent) or heat-inactivated serum (HIS, inactivated complement) in the presence or absence of 1 μg/ml of anti-human CD3 Ab (eBioscience, San Diego, CA) or 10 μg/ml of bacterial LPS (Sigma-Aldrich, St. Louis, MO) at 37°C for 30 min. After washing twice with phosphate-buffered saline (PBS), cells were subjected to PI staining for flow cytometric analysis (FACS) of cytolysis. To analyze complement-mediated virolysis, culture supernatants from NL4-3-infected Jurkat-7 or Jurkat cells were collected on day 20 postinfection. The supernatants were subjected to HIV-1 titration using the p24 ELISA assay (XpressBio, Thurmont, MD). The same amount of p24-containing HIV-1 particles from each cell line infected was incubated with the same volume of NHS or HIS in the presence or absence of 20 μg/ml of anti-HIV-1 gp120 mAb (2G12) or 10 μg/ml of LPS at 37°C for 30 min. After incubation, complement-mediated virolysis was quantitated by the amount of p24 released from lysed HIV-1 particles as previously described. 9 The measurement of p24 ELISA procedure was the same as the manufacturer's description, except that the lysis buffer was not used. As a consequence, we measured only the p24 released from the lysed viral particles triggered by the complement-mediated virolysis. The p24 in the core of the intact HIV-1 virions was not detected; therefore, p24 release served a parameter of virolysis. HIV-1 virions were treated with Triton X-100 for determination of total virolysis. The percentage of virolysis was calculated as follows: (p24 released in the presence of complement-competent serum − p24 released in the presence of HIS)/(p24 released from Triton X-100-treated virions − p24 released by medium only) × 100%. Mean ± standard deviation (SD) of three experiments was compared using the paired two-tailed Student t test.
HIV-1 binding, entry, and infection assays
HIV-1 NL4-3 at 20 ng/ml of p24 was incubated with 1 × 106 Jurkat or Jurkat-7 cells at the virus binding or entry conditions as previously described. 42 HIV-1 binding was conducted at 4°C for 30 min, while HIV-1 entry was carried out at 37°C for 2 h. After extensive washings with PBS, cells were subjected to extraction of total RNA for one-step RT-PCR (Qiagen, Valencia, CA) to measure viral RNA as previously described. 43 In some experiments, cells incubated with HIV-1 virions at 37°C for 2 h were treated with 0.05% trypsin (Sigma-Aldrich), followed by additional washings to remove noninternalized surface-bound virions. Cells were subjected to extraction of total RNA for one-step RT-PCR to evaluate HIV-1 entry. To compare the capacities of Jurkat-7 versus Jurkat cells in the production of HIV-1, NL4-3 particles (20 ng/ml of p24) were incubated with Jurkat-7 and Jurkat cells at 37°C for 2 h. After incubation, cells were washed twice with the culture medium and seeded in a six-well tissue culture plate with complete RPMI 1640 medium. Virus production in culture supernatants from the infected cells was monitored every other day during an infection course of 20 days using the p24 ELISA kit (XpressBio) as per manufacturers' instruction. In some experiments, total RNA and total DNA from infected cells were isolated and subjected to one-step RT-PCR or conventional polymerase chain reaction (PCR) assay to estimate intracellular HIV-1 production or replication.
TZM-bl cells were also used to test infectivity of HIV-1 virions from infected Jurkat and Jurkat-7 cells. Briefly, HIV-1 virions from supernatants of NL4-3-infected Jurkat and Jurkat-7 cells were used at the same amount of p24 to infect TZM-bl cells for 2 days. After infection, TZM-bl cells were lysed with the passive lysis buffer (Promega, Madison, WI) and subjected to measurement of luminescence (RLU) using the Dual-Luciferase Reporter Assay System (Promega). Supernatant samples containing higher quantities of infectious HIV-1 virions will have higher corresponding RLU values.
Semiquantitative RT-PCR and PCR assays
Total RNA was isolated from NL4-3-challenged cells using the TRIzol reagent (Invitrogen, Carlsbad, CA). The RNA samples were subjected to one-step RT-PCR using the reagents from Qiagen to detect HIV-1 Gag versus human housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as previously described. 43 HIV-1 Gag-specific primers include 5′-GGCAAGAGTTTTGGCTGAAG-3′ (forward) and 5′-CACATTTCCAACAGCCCTTT-3′ (reverse), while GAPDH-specific primers include 5′-GGTGGTCTCCTCTGACTTCAACA-3′ (forward) and 5′-GTTGCTGTAGCCAAATTCGTTGT-3′ (reverse). To detect viral DNA, total DNA, including viral double-stranded DNA (dsDNA) intermediates, proviral DNA, and cellular genomic DNA were extracted from the infected cells using the DNeasy DNA Isolation Kit (Qiagen). Isolated DNA was subjected to a conventional PCR using above-mentioned Gag primers to detect viral dsDNA intermediates and integrated DNA (provirus).
Fluorescent dyes, antibodies, and flow cytometric analysis (FACS)
Proaerolysin, a prototoxin of aerolysin, specifically binds to GPI anchors in the plasma membrane of cells. FLAER has been widely used in FACS to quantitate GPI-APs on the cell surface. 44 FLAER labeled with Alexa Flour 488 was purchased from Invitrogen (Grand Island, NY). Cholera toxin subunit B (CTB) conjugated with Alexa Fluor 488 or Alexa Fluor 647 was purchased from Invitrogen (Grand Island, NY). PI, PerCP-Cy5.5-conjugated anti-human CD4 mAb, PE-conjugated anti-human CD195 (CCR5), and mouse IgG1 conjugated with FITC were purchased from BD Pharmingen (San Diego, CA). BRIC229, a mouse anti-human CD59 mAb and FITC-conjugated anti-human CD59 mAb (MEM-43) were purchased from IBGR (Bristol, United Kingdom) and Thermo Scientific (Rockford, IL), respectively. Mouse anti-human CXCR4 (DyLight 650) mAb was from Thermo Scientific. Human anti-HIV-1 gp120 mAb (2G12) was purchased from Polymun Scientific Immunobiologische Forschung Gmbh (Klosterneuburg, Austria). All other secondary Abs conjugated with various fluorescent dyes were purchased from Invitrogen (Grand Island, NY).
To analyze the expression of total GPI-APs or CD59 on the cell surface, approximately 0.5 × 106 Jurkat-7 or Jurkat cells were incubated with FLAER (1:20 diluted in PBS) or FITC-conjugated anti-human CD59 mAb at room temperature for 20 min. Cells were washed twice with 2% FBS/PBS buffer, fixed with 1% paraformaldehyde (PFA) for 30 min, and subjected to FACS using FACSCaliber (BD Biosciences, San Jose, CA). In the parallel tests, FITC-conjugated isotype-matched mouse IgG was used as a background control. To analyze intracellular CD59 expression, cells were fixed and permeabilized with Cytofix/Cytoperm buffer (BD Pharmingen) at room temperature for 30 min before staining with FITC-conjugated anti-human CD59 mAb as previously described. 43 After washing, cells were subjected to FACS.
FACS data of both CSS and ICS were analyzed using FlowJo software (Tree Star, San Carlos, CA).
Restoration of GPI anchors in Jurkat-7 cells
Total RNA was isolated from Jurkat cells for complementary DNA (cDNA) synthesis using reverse transcriptase Superscript II and the oligo(dT)18 primer (Invitrogen, Grand Island, NY). The coding region, including all the exons of human PIG-A1 (genome variant 1), was amplified from the cDNA using the following primers: 5′-CCGTCTCAGCATGGCCTGTAGAGGA-3′ (forward) and 5′-AGGCTTCCTTCTACCTGGTTTCAG-3′ (reverse). PCR products were purified and cloned into the pcDNA 3.3-TOPO mammalian expression vector (pcDNA3.3-TOPO TA cloning kit; Invitrogen, Grand Island, NY). After expansion in E. coli (DH5α), pcDNA3.3 plasmid inserted with PIG-A1 (pPIG-A1) was confirmed by DNA sequencing. Jurkat-7 cells were transfected with pPIG-A1 or parental pcDNA3.3 plasmid as a control (pCtrl) using the Nucleofector Kit for Cell Lines and Amaxa Nucleofector devices (Lonza, Walkersville, MD) as per manufacturer's instruction. 37 After incubation for 24 h, transfected cells were subjected to either infection with NL4-3 to determine the cell sensitivity to HIV-1 infection or staining with FLAER and FITC-conjugated anti-human CD59 mAb to determine cell surface expression of GPI anchors and CD59, respectively.
Confocal microscopy
Jurkat and Jurkat-7 cells infected or uninfected with HIV-1 NL4-3, and Jurkat-7 cells transfected with pPIG-A1 or pCtrl were incubated with CTB conjugated with Alexa Fluor 647 (Life Technologies, Carlsbad, CA) at 4°C for 20 min to stain lipid rafts. After washing, these cells were incubated with mouse anti-human CD59 mAb (BRIC229) and human anti-HIV-1 gp120 mAb (2G12) at room temperature for 20 min, followed by staining with Alexa Fluor 546 (red)-conjugated anti-mouse and Alexa Fluor 633 (magenta)-conjugated anti-human secondary mAbs to stain CD59 and HIV-1 gp120, respectively. After washing, cells were resuspended in 150 μl of 1% PFA and mounted onto glass slides using the ProLong Gold Antifade reagent (Life Technologies, Carlsbad, CA) containing 4′,6-diamidino-2-phenylindole (DAPI) dye for fluorescent staining of DNA content and nuclei. Cells were analyzed using an Olympus FV1000-MPE confocal/multiphoton microscope fitted with a 603 objective. Signals were sequentially collected using single fluorescence excitation and acquisition settings to avoid crossover. 45 –47
Images were processed and analyzed using FV10-ASW 3.0 Viewer software (Olympus America, Center Valley, PA) or ImageJ 1.44p (NIH, Bethesda, MD).
Western blot
Jurkat and Jurkat-7 cells infected or uninfected with HIV-1 NL4-3 were lysed in 1 × cell lysis buffer (Cell Signaling Technology, Danvers, MA) plus 1 × protease inhibitors cocktail (Sigma-Aldrich), followed by protein extraction for Western blot analysis as previously described. 8,43 Isolated proteins were subjected to NuPAGE Novex gel electrophoresis (Invitrogen, Carlsbad, CA). Separated proteins were transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). After blocking with 5% non-fat skim milk in the TBST buffer at 4°C overnight, the membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-HIV-1 p24 Ab at room temperature for 1 h. After washing, the blot was visualized with an ECL detection system (Pierce, Rockford, IL). To prepare HIV-1 viral lysates, large volume of supernatants from NL4-3-infected Jurkat versus Jurkat-7 cells on day 20 postinfection was collected and subjected to virus concentration using an ultracentrifugation with 20% sucrose cushion at 35,000 rpm for 2.5 h. Virus pellets were harvested and lysed with the NuPAGE LDS sample buffer (Invitrogen, Grand Island, NY). Viral lysates were subjected to Western blot analysis of CD59 presence to determine human CD59 incorporation. Mouse anti-human CD59 mAb (BRIC 229) and an appropriate HRP-conjugated Ab were used as primary and secondary detection Abs, respectively. Blot was visualized with ECL detection system.
Statistical analysis
The paired two-tailed Student's t test was used to compare the mean ± SD. Values of p < .05 were judged significant.
Results
Jurkat-7 cells express neither GPI anchors nor CD59 on the cell surface
Jurkat-7 cells are deficient of GPI anchor expression on the cell surface due to the mutation of the PIG-A gene. 37,48 To confirm the results from these reports, we analyzed the presence of GPI anchors on the surface of Jurkat-7 cells using fluorescein-labeled proaerolysin (FLAER) that specifically binds to GPI anchors. 44 As shown in Figure 1A, the vast majority (>97%) of Jurkat-7 cells were negative for GPI anchors, whereas 100% of Jurkat cells were positive for GPI anchors. To test whether the deficiency of GPI anchors leads to a defect in the expression of CD59, one of many human GPI-APs, on the cell surface, we did cell surface staining (CSS) with a monoclonal antibody (mAb) (BRIC229) against human CD59. The results were similar to that of the FLAER staining. The majority (>95%) of Jurkat-7 cells did not express CD59 on the cell surface, whereas 100% of Jurkat cells were CD59 positive (Fig. 1B, middle column). We also performed intracellular staining (ICS) with BRIC229 and found that Jurkat-7 cells actually expressed intracellular CD59 at levels comparable to Jurkat cells (Fig. 1B, right column), indicating that CD59 deficiency on the cell surface is due to the lack of GPI anchors to anchor CD59 molecules to the cytoplasmic membrane. To confirm the flow cytometric analysis (FACS) data of CD59 expression on the cell surface and study cell membrane integrity, we used a confocal microscopy assay to compare CD59 expression and the appearance of lipid rafts on the surface of Jurkat-7 and Jurkat cells. Confocal microscopy analysis confirmed our FACS data that Jurkat-7 cells were CD59 negative, whereas Jurkat cells were CD59 positive (Fig. 1C, CD59 panel). We found no appreciable differences in the appearance of lipid rafts on the cell surface of Jurkat-7 versus Jurkat cells (Fig. 1C, CTB panel), suggesting that GPI anchor deficiency has no profound effect on the membrane organization. We also found that CD59 was strongly colocalized with cell membrane lipid rafts of Jurkat cells (Fig. 1C, Merge panel). Therefore, deficiency of GPI anchors in Jurkat-7 cells impairs expression of GPI-APs such as CD59, but has no effect on the appearance of lipid rafts and plasma membrane organization.

Characterization of intracellular and surface expressions of GPI anchor and CD59 by Jurkat versus Jurkat-7 cells.
Deficiency of GPI-APs on the surface of Jurkat-7 cells predisposes the cells to complement attack
CD59 is a key member of RCA and plays the most critical role in restricting complement activation as CD59 controls MAC formation at the terminal stage of the complement activation cascade through all three activation pathways. 14,15 We reasoned that the deficiency of CD59 on the surface of Jurkat-7 cells would predispose the cells to complement attack. To test this reasoning, we compared cytolysis efficacy of Jurkat-7 versus Jurkat cells in response to ADCML through the classical pathway of complement activation or lipopolysaccharide (LPS)-mediated alternative pathway activation. We found that wild-type Jurkat cells resisted complement attack in both conditions of ADCML and LPS-mediated CML as propidium iodide (PI)-positive Jurkat cells did not significantly increase when compared with that in the conditions of heat-inactivated complement (Fig. 2A, B). In contrast, Jurkat-7 cells were sensitive to complement attack as PI-positive Jurkat-7 cells dramatically increased in both conditions of ADCML and LPS-mediated CML (Fig. 2A, B). Notably, Jurkat-7 cells, but not Jurkat cells, were also sensitive to sera in the absence of Abs or LPS (Fig. 2B), indicating that sera may contain activated complement. Thus, deficiency of GPI anchors predisposes cells sensitive to complement attack through classical and alternative pathways of complement activation.
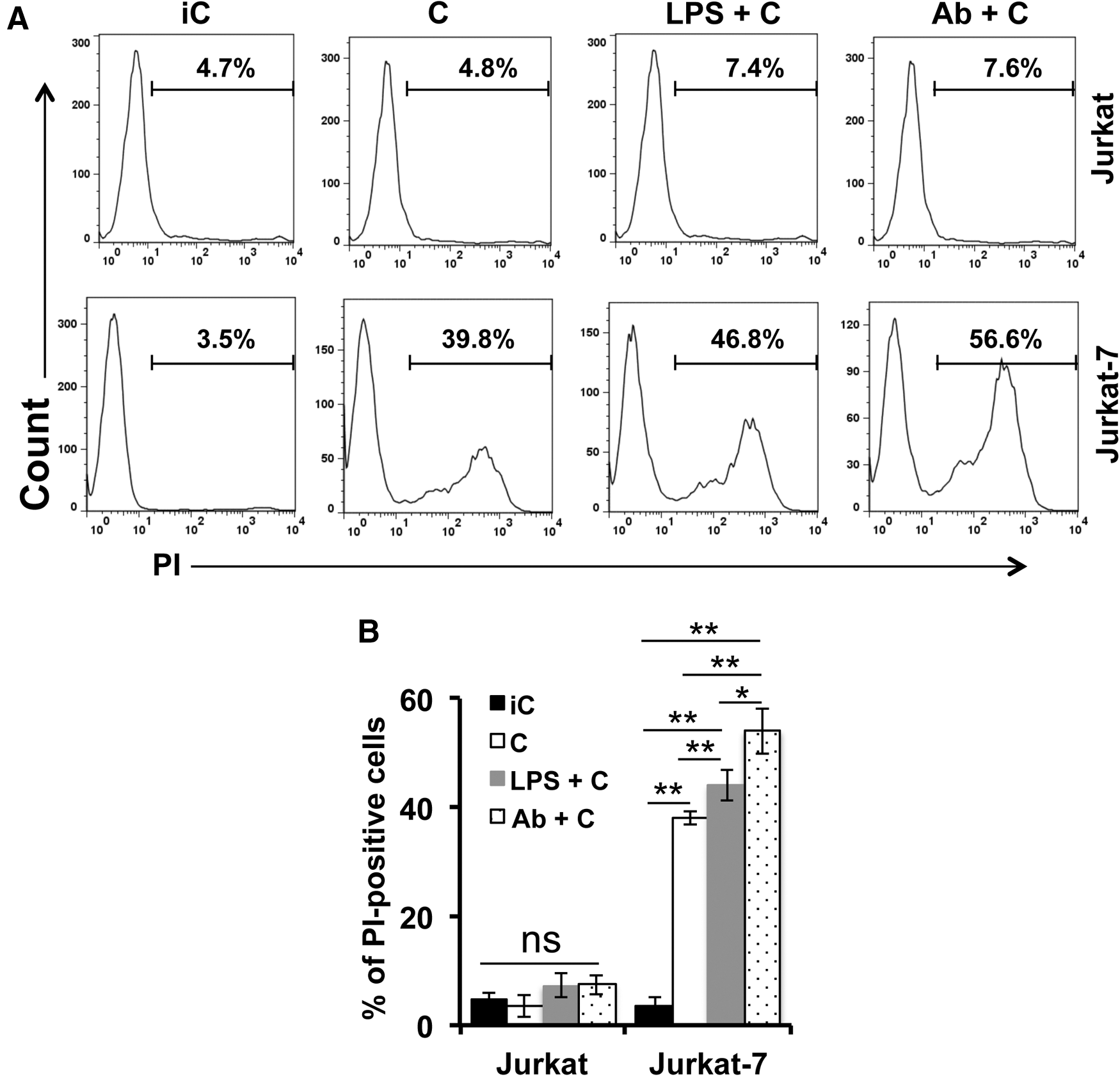
GPI anchor–deficient Jurkat-7 cells were sensitive to complement attack. Jurkat and Jurkat-7 cells were treated with C (complement competent) or iC (inactivated complement) in the presence or absence of anti-human CD3 Ab or LPS. Cells were subjected to PI staining to determine cytolysis.
GPI anchor deficiency attenuates the production of infectious HIV-1
Studies have demonstrated that Jurkat cells and GPI anchor–deficient Jurkat-7 cells have no significant differences in their morphology, proliferative capacity, and survival. 37 However, GPI anchor–deficient Jurkat-7 cells are more susceptible to Fas-mediated apoptosis than GPI anchor–positive parental cells, 37 indicating that GPI-APs on the cell surface are involved in cell signaling. It is unclear whether the cell signaling difference between Jurkat and GPI anchor–deficient Jurkat-7 cells reflects functional alterations of their intracellular compartments, thereby affecting HIV-1 replication. To clarify this issue, we used HIV-1 NL4-3 to infect Jurkat and Jurkat-7 cells and monitored the course of HIV-1 replication in these cells by determination of HIV-1 p24 concentrations in the cell culture supernatants. As shown in Figure 3A, the production of HIV-1 particles released from infected Jurkat cells markedly increased in a time-dependent manner during the course of 20 days, reaching 18 ng/ml on day 20 postinfection. In contrast, infected Jurkat-7 cells produced low, but detectable levels of HIV-1 over the course of 20 days, reaching 2 ng/ml on day 20 postinfection. These results were confirmed by p24 ICS. As shown in Figure 3B, more than 36% of Jurkat cells were p24 positive, whereas ∼2% of Jurkat-7 cells were p24 positive on day 20 postinfection.
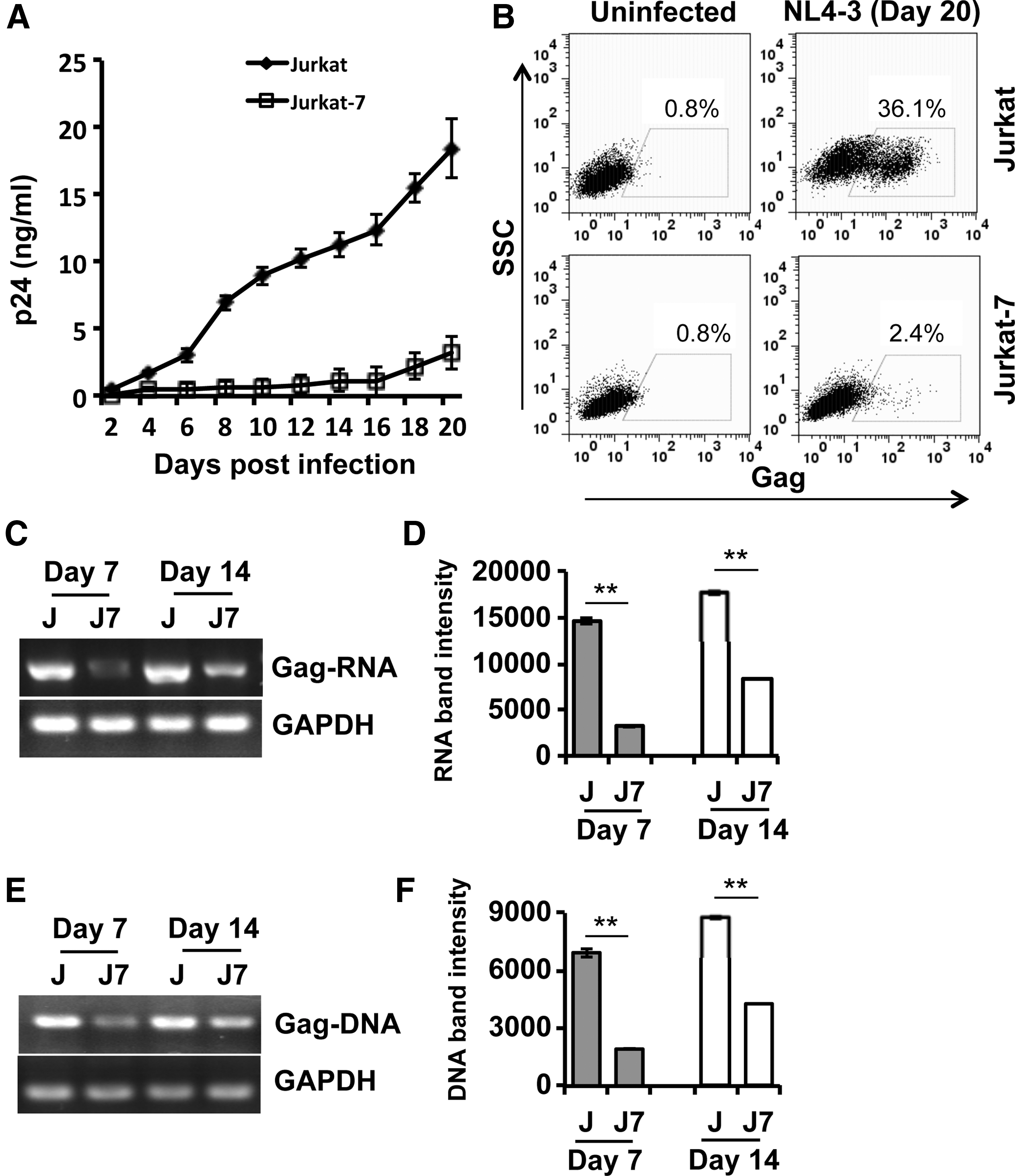
GPI anchor–deficient Jurkat-7 cells were less supportive to HIV-1 replication.
To understand which steps of the HIV-1 life cycle were affected by GPI anchor deficiency, we compared the amount of HIV-1 RNA (Fig. 3C, D) and DNA (Fig. 3E, F) in Jurkat versus Jurkat-7 cells. We found that the amounts of viral Gag messenger RNA (mRNA) and DNA were abundant and increased in a time-dependent manner in Jurkat cells during an infection course of 14 days (Fig. 3C–F). In contrast, infected Jurkat-7 cells produced Gag mRNA and DNA at low, but detectable levels (Fig. 3C–F). These results suggest that GPI anchor deficiency affects the HIV-1 life cycle.
GPI deficiency does not affect HIV-1 binding and entry into host cells
Viral binding and entry are the earliest stages of infection in the viral life cycle. HIV-1 binds to CD4, its primary receptor, and a coreceptor (CCR5 or CXCR4) to initiate viral entry and infection. We compared the efficacy of HIV-1 binding and entry into Jurkat versus Jurkat-7 cells to investigate whether these two steps were affected by GPI anchor deficiency. To study HIV-1 binding, Jurkat and Jurkat-7 cells were incubated with NL4-3 at a multiplicity of infection (MOI) of 5 at 4°C for 30 min. After washing extensively to remove unbound viral particles, cells were subjected to RNA isolation for RT-PCR to detect viral RNA abundance. As shown in Figure 4A, comparable amounts of viral RNA were detected in Jurkat versus Jurkat-7 cells, indicating that GPI anchor deficiency does not affect HIV-1 binding to host cells. Since the binding efficacy of HIV-1 particles is predominantly determined by the abundance of functional HIV-1 receptor and coreceptor molecules on the cell surface, we measured the levels of CD4 and chemokine receptor molecules that serve as the HIV-1 primary receptor and coreceptors, respectively. We found that Jurkat-7 cells expressed a similar density of CD4, CCR5, and CXCR4 molecules on the cell surface to that on the surface of Jurkat cells (Fig. 4B), indicating that GPI anchor deficiency has no effects on the expression of CD4 molecules and chemokine receptors on the cell surface. This result is understandable because they are not GPI-APs, as CD4 is a member of the immunoglobulin superfamily, while the chemokine receptors of CCR5 and CXCR4 are seven-transmembrane proteins.
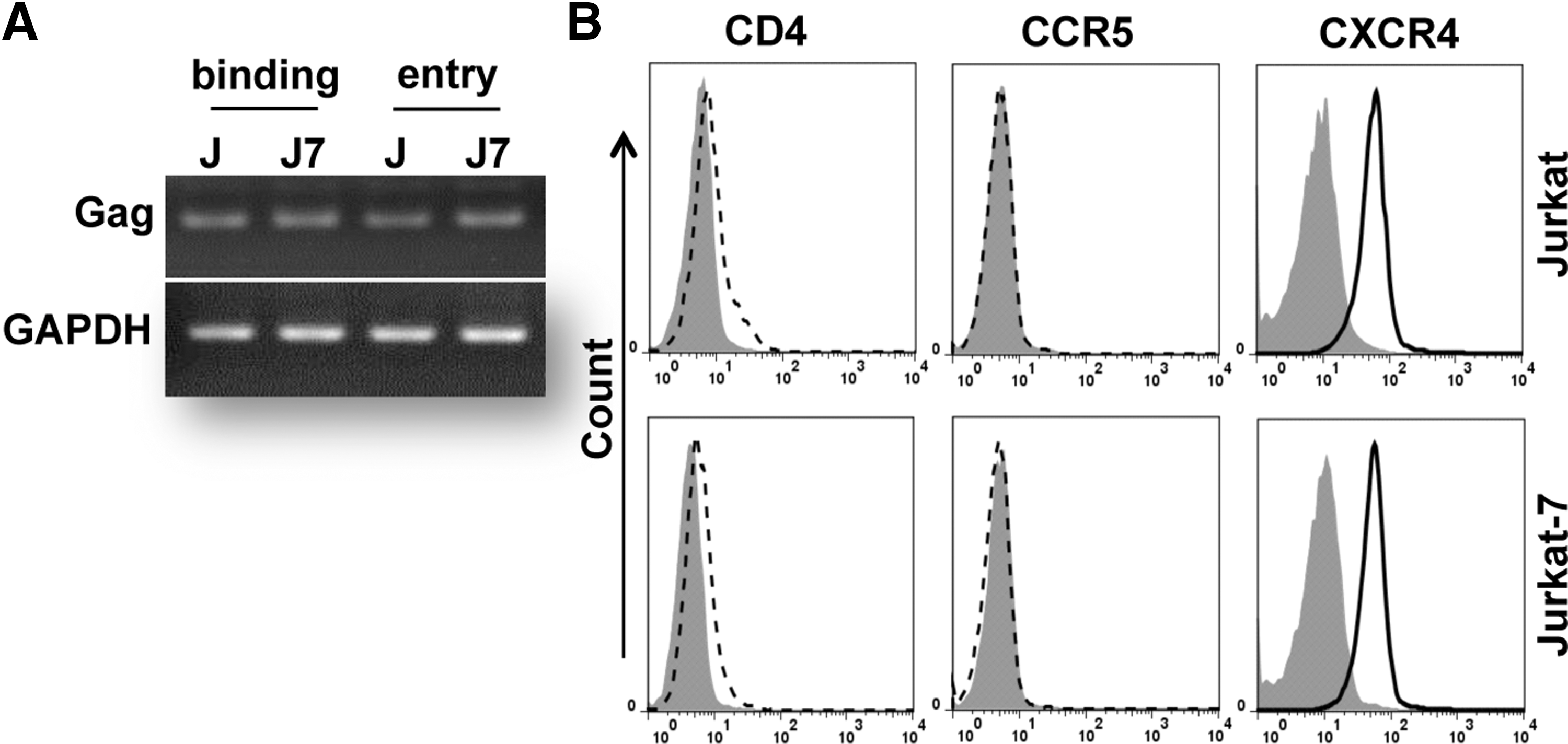
GPI anchor deficiency did not affect HIV-1 binding and entry into the host cells.
HIV-1 binding triggers rearrangements of the cytoplasmic membrane to allow virus entry into target cells. Following analysis of virus binding, we studied the effects of GPI anchor deficiency on virus entry. Jurkat-7 and Jurkat cells were incubated with NL4-3 at 37°C for 2 h, a condition that allows HIV-1 particles to complete the entry process. Cells were subjected to RNA isolation for RT-PCR to detect viral RNA abundance. As shown in Figure 4A, comparable amounts of viral RNA were observed in Jurkat versus Jurkat-7 cells, and these results were not affected by trypsin treatment that removes noninternalized surface-bound virions (data not shown), indicating that GPI anchor deficiency does not affect HIV-1 entry into target cells.
HIV-1 virions produced from GPI anchor–deficient cells are less infectious and more sensitive to complement attack
HIV-1 virions selectively acquire more than 40 host proteins from infected cells. 30 Among these host proteins, two RCA members of GPI-APs, CD55 and CD59, have been identified at biologically functional levels on the surface of HIV-1 particles and provide vital protection to HIV-1 particles against complement attack. They may also be required to maintain virion structure and infectivity. To test the possibility, we used the same p24 amount of HIV-1 virions prepared from infected Jurkat and Jurkat-7 cells to infect Jurkat cells. The production of virus particles in the culture supernatants was monitored every other day for 12 days. As shown in Figure 5A, Jurkat cells infected with Jurkat-derived HIV-1 produced high levels of virions in the course of 12 days of infection. In contrast, Jurkat cells infected with Jurkat-7-derived HIV-1 produced detectable, but low levels of virions in the course of 12 days of infection. Similar results were obtained for HIV-1 infection of TZM-bl cells. As shown in Figure 5B, TZM-bl cells infected with Jurkat-derived HIV-1 produced higher corresponding relative luciferase unit (RLU) values than the cells infected with Jurkat-7-derived HIV-1. Reduction of Jurkat-7-derived HIV-1 infectivity is, at least, partly due to a reduced activity of virus binding as Jurkat-7-derived HIV-1 exhibited relatively weak binding to wild-type Jurkat cells (Fig. 5C). These results indicate that virions produced from Jurkat-7 cells are defective and less infectious.

Virions from infected Jurkat-7 cells were less infectious, CD59 defective, and vulnerable to complement attack.
We also collected large amounts of cell-free supernatants from HIV-1-infected Jurkat and Jurkat-7 cells on day 20 postinfection to concentrate and purify virus particles by ultracentrifugation. These virus particles were subjected to Western blot to detect incorporated CD59. HIV-1 p24 was also detected as a virus loading control. As expected, a high level of CD59 was detected in virions from the supernatant of infected Jurkat cells, while little or no CD59 was detected in virions from the supernatant of infected Jurkat-7 cells (Fig. 5D). The HIV-1 p24 intensity is comparable in supernatant virions from infected Jurkat versus Jurkat-7 cells (Fig. 5D), indicating that the same amount of virus particles was used in our experiments. Our results provide evidence that HIV-1 virions incorporate host proteins from infected cells, and the virions most likely acquire RCA members from the surface of infected cells.
Lack of RCA members, including CD59, may predispose HIV-1 virions to complement attack. To test this reasoning, we analyzed virolysis of HIV-1 virions from infected Jurkat and Jurkat-7 cells in response to CML through classical and alternative pathways of complement activation. Complement plus anti-HIV-1 gp120 mAb (2G12) was used to trigger complement activation through the classical pathway, while LPS plus complement was used to trigger complement activation through the alternative pathway. We found that HIV-1 virions from infected Jurkat-7 cells were sensitive to CML through both classical and alternative pathways of complement activation, whereas HIV-1 virions from infected Jurkat cells weakly responded to complement attack through either classical or alternative pathways of complement activation (Fig. 5E). Thus, HIV-1 virions that lack RCA members, including CD59, are vulnerable to CML through both classical and alternative pathways of complement activation.
Restoration of GPI anchors enables Jurkat-7 cells to express cell surface CD59 that is colocalized with HIV-1 gp120/gp41 within lipid rafts after virus infection
We next studied whether CD59 was expressed on the cell surface after GPI-anchor restoration and whether CD59 and HIV-1 gp120/gp41 were colocalized on the surface of GPI anchor–restored Jurkat-7 cells. Jurkat-7 cells are a GPI anchor–deficient derivative mutant of Jurkat cells due to a PIG-A1 gene mutation. 37 We therefore cloned the parental PIG-A1 gene from Jurkat cells into a pcDNA3.3-TOPO mammalian expression vector and transfected the PIG-A1-expressing plasmid construct (pPIG-A1) into Jurkat-7 cells to restore GPI anchor function. FACS analysis of GPI anchors and CD59 on the cell surface revealed that Jurkat-7 cells transfected with pPIG-A1, but not the control plasmid construct (pCtrl), expressed high levels of both GPI anchors and CD59 (Fig. 6A). We also used a confocal microscopy assay to analyze CD59 and gp120/gp41 colocalization on the surface of pPIG-A1-transfected Jurkat-7 cells after infection with HIV-1 for 24 h. The confocal microscopy analysis confirmed our FACS data that pPIG-A1-transfected Jurkat-7 cells expressed CD59 on the surface (Fig. 6B). As we previously reported, 49 CD59 on the cell surface was strongly associated with cell membrane lipid rafts (Fig. 6B, yellow in Merge panel). We found a similar association between gp120/gp41 and lipid rafts on the surface of infected Jurkat-7 cells that were transfected with the pPIG-A1 plasmid construct, but not pCtrl plasmid construct (Fig. 6C). Importantly, CD59 and gp120/gp41 were colocalized in lipid rafts on the surface of infected Jurkat-7 cells that were transfected with the pPIG-A1 plasmid construct (Fig. 6C, yellow in Merge panel), suggesting that gp120/gp41 may directly interact with RCA molecules on the cell surface. Our results suggest that CD59 is located in close proximity to viral proteins such as gp120/gp41 on the cell surface, facilitating direct interaction between them.
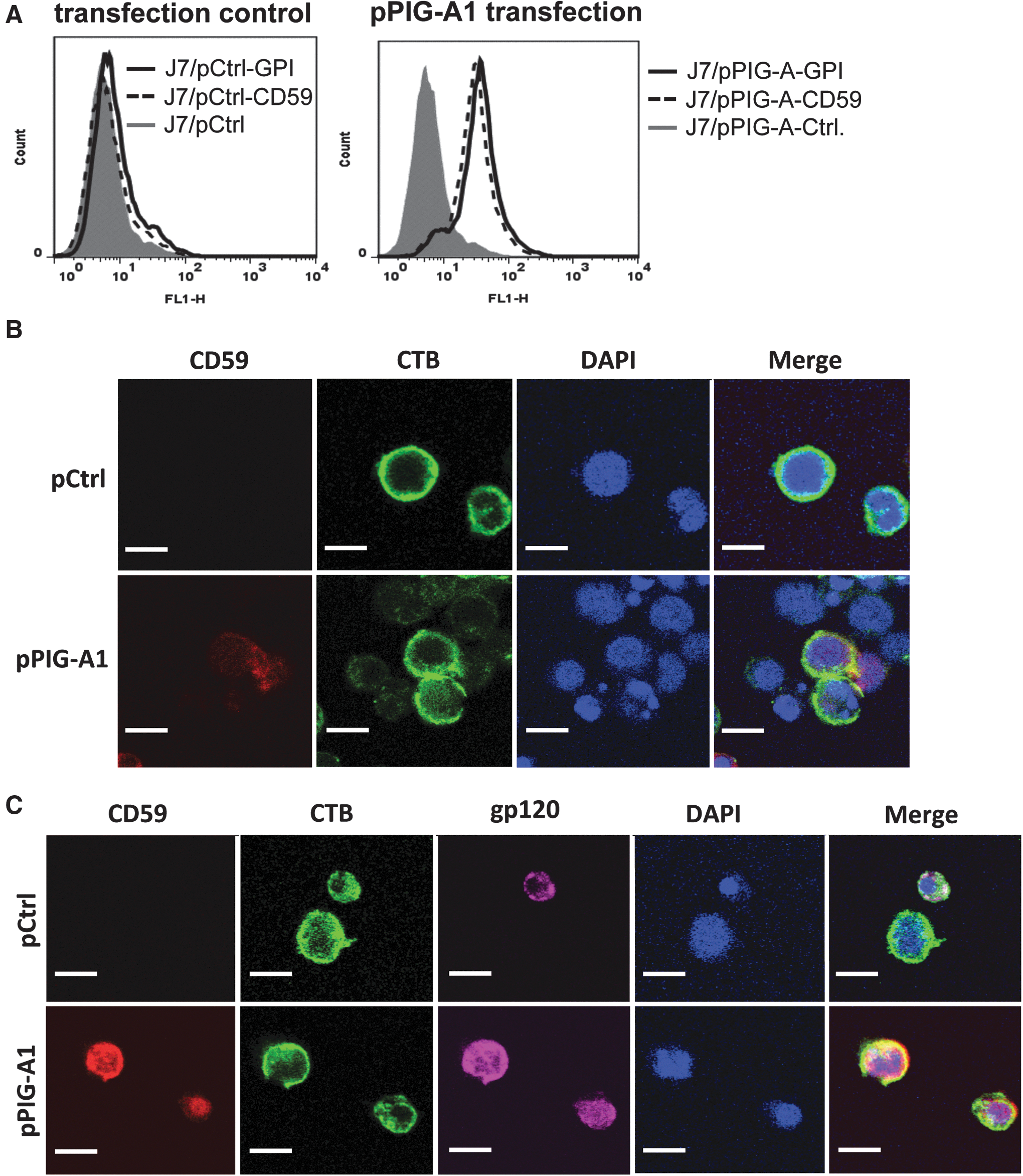
Restoration of GPI anchor biosynthesis rendered CD59 expression on the surface of Jurkat-7 cells.
Discussion
Previous studies reported by our group and others have demonstrated that HIV-1 incorporates host cell proteins, including RCA members, into its envelope to escape complement attack. 9 –14 It is believed that HIV-1 acquires biologically functional RCA members such as CD59 during viral budding at the plasma membrane of infected cells. We used GPI anchor–deficient Jurkat-7 cells that lack CD59 on the cell surface as a model to study the role of GPI-APs, particularly CD59, in the HIV-1 life cycle, incorporation of host proteins into virions, and CML of virions. FACS data of intracellular versus cell surface GPI-APs confirmed that Jurkat-7 cells expressed neither GPI anchor nor CD59 on the cell surface (Fig. 1A, B), but expressed intracellular CD59 at levels comparable to that in the parental Jurkat cells (Fig. 1B), indicating that CD59 deficiency on the cell surface is due to lack of GPI anchors to anchor CD59 molecules into the cytoplasmic membrane. Similarly, we also found that Jurkat-7 cells only expressed intracellular bone marrow stromal cell antigen 2 (BST-2, a member of GPI-APs), whereas Jurkat cells expressed both intracellular and surface BST-2 (data not shown). In fact, the data were validated by our results that transfection of Jurkat-7 cells with PIG-A1-expressing plasmid constructs restored the expression of the total GPI-APs and CD59 on the cell surface (Fig. 6). Since Jurkat-7 cells expressed intracellular CD59, but not surface CD59, we analyzed CD59 incorporation in viral particles from the cell-free supernatants of HIV-1-infected Jurkat-7 cells to determine where CD59 incorporation into virions occurred. As expected, a high level of CD59 was detected on virions from the supernatant of infected Jurkat cells, while little or no CD59 was detected on virions from the supernatant of infected Jurkat-7 cells (Fig. 5C). HIV-1 p24 intensity was comparable in supernatant virions from infected Jurkat versus Jurkat-7 cells (Fig. 5C), indicating that the same amount of virus particles was used in our experiments. Our results provide evidences that HIV-1 virions are most likely to acquire CD59, and probably other host proteins, from the surface of infected cells, and not from intracellular compartments.
Both HIV-1-infected cells and virions use their surface RCA members, including CD55 and CD59, to resist ADCML. 1,9 –13,49 Abrogation of the biological function of RCA members, particularly CD59 (a key RCA member that controls formation of the MAC at the terminal stage of the complement activation cascade through all three activation pathways), 14,15 renders both HIV-1-infected cells and virions sensitive to ADCML. 1,9 –13,49 In agreement with these findings, our experiments showed that Jurkat-7 cells and HIV-1 particles from infected Jurkat-7 cells were sensitive to ADCML in response to treatment with anti-human CD3 and anti-gp120 mAb 2G12 when compared with parental Jurkat cells and viral particles from infected Jurkat cells, respectively (Figs. 2A, B, and 5D). In addition, Jurkat-7 cells and HIV-1 particles from infected Jurkat-7 cells were also sensitive to CML through the alternative pathway of complement activation, as they were effectively lysed by LPS-triggered complement activation (Figs. 2A, B, and 5D). Thus, HIV-1 virions and infected cells use RCA members to protect themselves from adaptive and innate immune responses by suppression of the classical and alternative pathways of complement activation. In HIV-1-infected patients, anti-gp120 Abs are vigorously elicited, but fail to complete ADCML against virions due to the presence of RCA on the virion surface. 1, 10,11 In addition to elicitation of virus-specific Abs, HIV-1-infected patients have significantly higher levels of circulating LPS due to increased bacterial translocation from the damaged gastrointestinal tract. These Abs and LPS present in the circulation of HIV-1-infected patients may activate the early complement cascade, but not fully complete complement activation as MAC pores are not formed, leading to viral and cellular escape. 28,29
GPI anchor deficiency also attenuates the production of infectious HIV-1. The production of HIV-1 particles released from infected parental Jurkat cells markedly increased in the infection course of 20 days (Fig. 3A). In contrast, infected Jurkat-7 cells produced low levels of HIV-1 in the same infection course (Fig. 3A). HIV-1 p24 ICS also showed that the percentage of p24-positive Jurkat-7 cells was much lower compared with p24-positive Jurkat cells (Fig. 3B). The reduction of HIV-1 virions in Jurkat-7 cells versus Jurkat cells was not due to a lower susceptibility of GPI anchor–deficient cells to HIV-1 binding and entry, as comparable amounts of cell-bound virions and virus-containing cells were detected in Jurkat-7 versus Jurkat cells (Fig. 4A). In addition, we found that GPI anchor deficiency had no effects on the surface expression of the HIV-1 receptor or coreceptors as Jurkat-7 cells expressed a similar amount of CD4, CCR5, and CXCR4 molecules on the cell surface to that on the surface of Jurkat cells (Fig. 4B). In infected cells, HIV-1 proteins, including Gag and glycoproteins, are matured or glycosylated in an intracellular apparatus such as the ER or Golgi. The glycoproteins are then cleaved into envelope membrane spanning (gp41) and extracellular subunits (gp120), which associate to form trimers and migrate to lipid rafts within the plasma membrane. Concordantly, GPI anchors are also synthesized in the cytoplasmic leaflet of the ER membrane bilayer, 50 and GPI-APs such as CD59 is located in the plasma membrane within lipid rafts. Thus, the interactions between GPI-APs and HIV-1 may occur in an intracellular apparatus such as the ER or in the lipid rafts of the plasma membrane. In fact, we found that the amounts of viral Gag mRNA and DNA were abundant and increased in a time-dependent manner in Jurkat cells during an infection course of 14 days (Fig. 3C–F). In contrast, infected Jurkat-7 cells produced Gag mRNA and DNA at low, but detectable levels (Fig. 3C–F). These results suggest that the synthesis of viral RNA and DNA was attenuated in GPI anchor–deficient cells. Taken together, GPI anchor deficiency attenuates the production of infectious HIV-1 through affecting the HIV-1 life cycle.
In conclusion, this study is the first report to demonstrate that HIV-1 virions incorporate RCA members such as CD59 from the cell surface rather than the intracellular compartments. GPI anchor deficiency attenuates the production of infectious HIV-1 by impairing the HIV-1 life cycle. Deficiency of GPI-APs on the cell surface and virions renders them sensitive to complement attack through both classical and alternative pathways of complement activation.
Footnotes
Acknowledgments
We thank Dr. J. Schubert at the Hannover Medical School (Germany) for providing Jurkat-7 cells. The following reagents were obtained through the National Institutes of Health AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases: HIV-1 NL4-3 strain, NL4-3 plasmid (pNL4-3), anti-HIV-1 gp120 mAb 2G12, and TZM-bl. This work was supported, in part, by a Grand Challenges Explorations Phase II (GCE II) grant through the Bill & Melinda Gates Foundation (OPP1035237 to Q.Y.), NIH Grants (R33AI104268 and R01AI117835 to Q.Y.), the Showalter Research Trust Fund (to Q.Y.), NIH Grant T32 AI060519 (to D.B.), and Research Facilities Improvement Program Grant C06 RR015481 from the National Center for Research Resources, NIH, to Indiana University School of Medicine.
Author Disclosure Statement
No competing financial interests exist.