
Abstract
Lenalidomide, an oral immunomodulating agent, has shown promising activity in HIV-infected individuals with Kaposi's sarcoma (KS). This single-arm, multicenter, open-label, Gehan's two-stage phase II trial evaluated the efficacy and safety of lenalidomide in HIV-infected patients with progressive KS despite previous chemotherapy (NCT01282047, ANRS 154 Lenakap trial). The primary endpoint was the rate of partial response (PR) or complete response (CR) at week 24, evaluated by both the study investigators and the patients using the Physical Global Assessment (PGA). AIDS Clinical Trials Group (ACTG) criteria for KS treatment evaluation were used as a secondary endpoint. The data and safety monitoring board recommended that enrollments be halted on April 24, 2013, because of lack of responses. We enrolled 12 antiretroviral-treated HIV-infected men with progressive KS despite previous chemotherapy. Their HIV plasma viral load was <50 copies/ml and their median CD4 cell count 444/mm3. One patient stopped taking lenalidomide because of hives at week 1 and a second patient died at week 7. The remaining 10 patients were assessable at week 24, when none had PGA-defined CR or PR and one had ACTG-defined PR. There were no additional PGA responses at week 48, but an additional three patients had ACTG responses, for a total of four patients with ACTG PR at week 48 (40%; 95% confidence interval: 12.2–73.8). Fourteen grade 3–4 adverse events were considered at least possibly related to lenalidomide during a total of 101 cycles. Lenalidomide was well tolerated in antiretroviral experienced patients with progressive KS previously treated with chemotherapy. The ACTG-defined response rate at week 48 was 40%, while it was 0% using PGA criteria.
Introduction
K
KS is due to human herpesvirus type 8 (HHV-8), which provokes angiogenesis and induces proinflammatory cytokine production (interleukin [IL]-1, IL-6, tumor necrosis factor [TNF]-α, and interferon (IFN)-γ). 5,6 cART and/or cytotoxic chemotherapy are the standard treatments for KS associated with HIV infection (HIV-KS), giving response rates of 20%–76% but troublesome adverse effects. 7 –13 Long-term chemotherapy is often necessary, resulting in cumulative toxicity, risk of drug–drug interactions, and negative impact on quality of life. There are no oral drugs approved in KS.
Thalidomide, an oral immunomodulating agent, has anti-TNF-α and antiangiogenic activities. Previous case reports and phase II studies showed inhibition of HHV-8 replication and KS regression in 34%–47% patients treated with thalidomide, 14 –18 but use of this drug is limited by its frequent adverse effects. Lenalidomide, a second-generation thalidomide derivative approved in patients with myelodysplastic syndromes and myeloma, is more potent and avoids many of the serious adverse effects, including neuropathy. 19 –23 Lenalidomide is an immunomodulatory drug with antiangiogenic, direct antitumoral, and immunostimulatory properties. 19 –28 It inhibits TNF-α, IL-1, IL-6, IL-12, and vascular endothelial growth factor (VEGF) expression and increases IL-8, IL-10, and IFN-γ production. 26,29 Reports of four cases of HIV-associated KS suggest that lenalidomide can induce tumor regression and is well tolerated, thus warranting prospective studies. 30 –32
In this study, we evaluated the efficacy and safety of lenalidomide in a multicenter, open-label, phase II trial in HIV-infected patients with previously treated KS and plasma viral load (pVL) <50 copies/ml (ANRS 154 Lenakap study).
Patients and Methods
Study design
This single-arm, multicenter, open-label, Gehan's two-stage phase II trial was designed to evaluate the efficacy and safety of lenalidomide after 24 weeks in patients with controlled HIV infection and progressive KS despite previous chemotherapy. The trial conformed to good clinical practices and to the Declaration of Helsinki. The Institutional Review Board of Paris Ile de France VII approved the study, and all the patients provided their written informed consent before participating. The study is registered in ClinicalTrials.gov (NCT01282047, ANRS 154 Lenakap).
Patients
The following patients were eligible: HIV-infected men and non-breastfeeding women with a negative βHCG test, aged between 18 and 75 years with progressive biopsy-proven KS, failure or relapse after one or more lines of anti-KS chemotherapy, HIV pVL <50 copies/ml whatever the CD4 cell count, ongoing cART for at least 12 months, and a Karnofsky performance status >70%. Men and women were informed on the teratogen risk of lenalidomide. Adequate contraception was required 4 weeks before starting lenalidomide and maintained up to 4 weeks after stopping lenalidomide. Check of childbearing potential contraception and pregnancy test (minimum sensitivity of 25 mIU/ml) was made at all visits during the follow-up. Eligible patients were required to respect wash-out periods of at least 4 weeks for specific KS chemotherapy and 8 weeks for IFN-α therapy.
Patients were not eligible if they had only visceral, cardiac, and/or bronchopulmonary KS, two pVL values >50 copies/ml during the past 6 months, ongoing infections, heart disease, Castleman's disease, cancers, previous or current hematological malignancies, grade >2 polyneuritis, alanine aminotransferase or aspartate aminotransferase >3 normal values, neurotoxic drug therapy, polymorphonuclear neutrophil count <1,000/mm3, platelet count <75,000/mm3, life expectancy less than 2 months, creatinine clearance ≤50 ml/min, concomitant antineoplastic drug therapy, known allergy or hypersensitivity to lenalidomide and/or thalidomide, or a contraindication to anticoagulation.
Study treatments
The recommended starting dose of lenalidomide was one 25 mg capsule on days 1–21 followed by a 7-day wash-out period every 28 days, repeated for 24 weeks. From week 24, lenalidomide was stopped in case of disease progression, and the patients were followed until week 48. Lenalidomide was to be continued for a further 12 weeks in case of complete response (CR) and for 24 weeks in case of partial response (PR) or stable disease. Lenalidomide was provided by Celgene (France) as 5 and 25 mg hard capsules.
As recommended in the lenalidomide summary of product characteristics in the granted market organizations, aspirin was given to prevent thromboembolic events. Anticoagulation at therapeutic doses was preferred to aspirin for patients with a history of pulmonary embolism or deep venous thrombosis, or with relevant risk factors. The lenalidomide dose could be adapted in case of skin rash, thrombocytopenia, renal impairment, or neutropenia (5, 10, or 15 mg). Granulocyte colony-stimulating factors could be prescribed in case of neutropenia.
Follow-up
After screening (less than 4 weeks before treatment initiation), study evaluations were performed at day 0 (baseline), week 1 and 4, and every 4 weeks thereafter until week 48. At each visit, the patients had a routine safety assessment, HIV pVL and CD4 cell measurements, and blood sampling. The total number of lesions was recorded at screening, and 4–6 index lesions were chosen to be evaluated by the study investigator and the patient at each visit, and retrospectively by the validation committee on digital photographs; the type and location of the index lesions were recorded. The procedure to take pictures was standardized as described in an annex of the protocol. The Physical Global Assessment (PGA) was used by both the study investigators and the patients to score the index lesions. The degree of infiltration and size of the index lesions were assessed by the study investigator. A 100% decrease in the PGA score for an index lesion was considered to represent CR, 51%–99% PR, 26%–50% a slight improvement, an increase or decrease of less than 25% stable disease, and an increase of 25% or more progression. The final response was evaluated on the basis of the index lesion with the poorest response. AIDS Clinical Trials Group (ACTG) criteria were also used. 33 ACTG criteria allow to define four outcomes: CR (no lesions for at least 4 weeks), PR (a decrease of at least 50% in the number and size of the lesions for at least 4 weeks, with no new lesions), progression (increase of at least 25% in the number and size of the lesions and/or appearance of a new lesion), and stable disease (all other situations).
HHV-8 viral load was determined at day 0, week 4, 12, and 24. HHV-8 genome detection was based on real-time polymerase chain reaction as described elsewhere. 34 Adverse events were recorded at each visit and graded with the ANRS scale (France Recherche Nord & Sud Sida-HIV Hépatites). The investigators evaluated the causal relationship with the study treatment.
Cytokine assays
Blood was collected in heparinized tubes at baseline and each month during the study. Plasma specimens were stored at −80°C until analysis. Plasma samples obtained at week 0, 24, and 48 were used for cytokine assays and pharmacokinetic studies.
Plasma concentrations of IFN-γ, TNF-α, VEGF-A, IL-1β, IL-4, IL-6, IL-8, IL-10, IL-12p70, and IL-17A were measured in duplicate with a multiplex electrochemiluminescence assay (V-Plex™ Custom Human Cytokine Assay; Meso-Scale Discovery) according to the manufacturer's protocol, using 25 μl of plasma from each time point. Data acquisition was performed by the Cochin Cytometry and Immunobiology Facility (Paris, France). The lower limits of quantification (LOQ) were 0.02 pg/ml for IL-4, 0.03 pg/ml for IL-10, 0.04 pg/ml for TNF-α, IL-1β, and IL-8, 0.06 pg/ml for IL-6, 0.11 pg/ml for IL-12p70, 0.20 pg/ml for IFN-γ, 0.74 pg/ml for IL-17A, and 1.12 pg/ml for VEGF-A. By convention, concentrations below the LOQ were defined as LOQ/2 for statistical analysis.
Pharmacokinetic study
Plasma concentrations of antiretroviral (ARV) drugs 35 and lenalidomide 36 were determined by using two validated ultrahigh-performance liquid chromatography methods coupled with tandem mass spectrometry (LC-MS/MS; Waters Acquity® UPLC and TQD, Milford, MA).
Non-nucleoside reverse transcriptase inhibitor, protease inhibitor, plasma trough concentrations (C12 h or C24 h) were considered adequate if they matched the concentrations proposed by the French national guidelines for antiretroviral therapy of HIV-1 infection in adults 37 : 1,000–4,000 ng/ml for efavirenz, 200 ng/ml for atazanavir, 2,000 ng/ml for darunavir, and 4,000 ng/ml for lopinavir. Based on published data, lenalidomide, 36 raltegravir, 38 and tenofovir 39 C24 h values were considered adequate if 3–20, 15, and 22–66 ng/ml, respectively.
Endpoints
The primary endpoint was the proportion of patients with a PR or CR at week 24, as evaluated by both the study investigators and the patients using the PGA score for the 4–6 index lesions. A validation committee judged each patient's outcome from all available photographs. In case of discordant judgments, the validation committee evaluation was retained.
Secondary endpoints included the proportion of patients with a PGA-defined PR or CR at week 48; the percentage change from baseline in the sum of the surface areas of the 4–6 index lesions at weeks 24 and 48 (ACTG criteria); the change from baseline in plasma HIV pVL, HHV-8 viral load, CD4 and CD8 cell count, CD4/CD8 ratio at week 48, hemoglobin level, creatinine clearance, platelet, leukocyte, and neutrophil counts, in cytokine and drug levels, KS-event-free survival at week 48 (an event was defined as disease progression or death), clinical and biological tolerability of lenalidomide, and treatment adherence (number of pills consumed divided by the number of pills received).
Statistical analysis
The sample size was chosen as in a typical Gehan's two-stage phase II trial. Fourteen assessable patients were required in the first step. If no response was observed in the 14 assessable patients, we could exclude, with a type 1 error of 5%, a response rate higher than 20%; if one response (PR or CR) was observed, other patients could be included up to a total of 25 assessable patients. Patients who discontinued the study before primary endpoint evaluation were replaced by other patients to reach the target sample size.
The Data Safety Monitoring Board recommended that inclusions in the study be prematurely discontinued on April 24, 2013, after 12 individuals had been enrolled, because of a lack of CR and PR. The independent committee specified that the a posteriori evaluation of lesions on photographs was difficult, linked to the quality of some photographs in the absence of standardized procedure to take it. The patients already included were followed until week 48 as originally planned.
The primary efficacy endpoint was assessed for all assessable patients. Patients who died of KS-unrelated causes before the response evaluation or who withdrew from the study were not assessable. Patients who prematurely discontinued the study treatment but remained in the study were included in the efficacy analysis. All patients who received at least one dose of study treatment were included in the safety analysis.
Data were summarized as proportions for categorical variables, and median/range for continuous variables. Changes in continuous variables between baseline and week 48 were compared using the Wilcoxon paired test, and the nonparametric Spearman correlation test was used to study associations between continuous variables. For cytokine parameters with at least one censored value due to the assay LOQ, we used the Kaplan–Meier method to estimate the changes between baseline and week 48. The Mann–Whitney nonparametric test was used to compare changes in cytokine levels between patients with progression and those with stable disease or improvement at week 48. The Kaplan–Meier method was used to estimate the progression-free survival rate. Events were defined as disease progression or death before week 48. Time to an event was defined as the time between the date of lenalidomide initiation and the date of the event. All reported p values are two tailed, and significance was assumed at p < .05. All analyses were performed with IBM SPSS statistics software version 22.0 for Windows (SPSS, Inc., Chicago, IL).
Results
Patient disposition and characteristics
We enrolled 12 men with HIV-related biopsy-proven KS among 15 patients screened in nine French hospitals between November 2011 and February 2013 (Fig. 1).
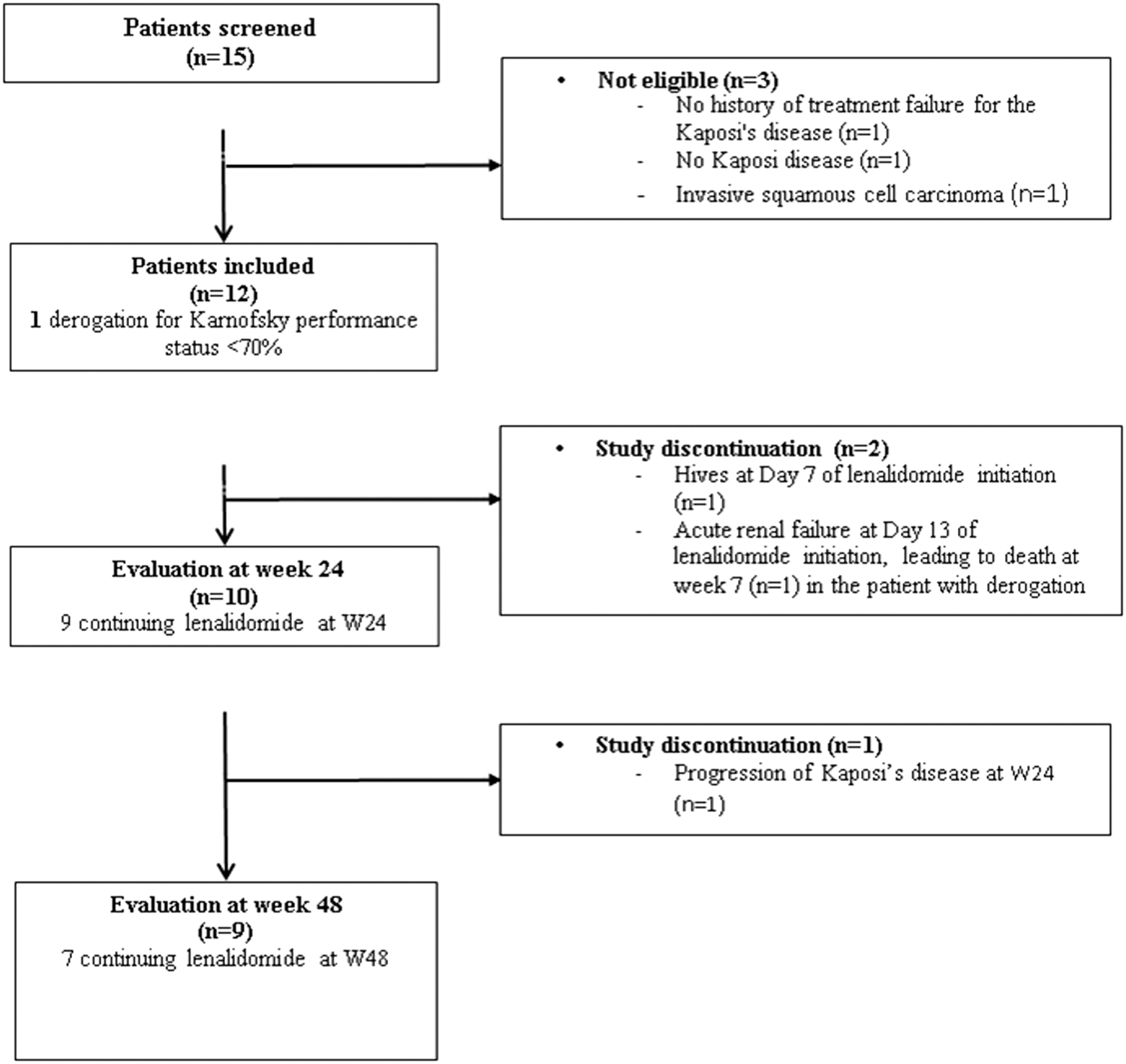
Study flowchart.
Baseline characteristics of the 12 patients are shown in Table 1. All the patients had cutaneous KS, with a median of 21 lesions, and one patient also had visceral involvement (gastrointestinal tract). The patients had received between 1 and 13 courses of previous chemotherapy regimens during a median period of 16 months. The most commonly used drug classes were anthracyclines (13 doxorubicin, 3 pegylated-doxorubicin, 5 daunorubicin), cytotoxic antibiotics (13 bleomycin), vinca alkaloids (3 vinblastine, 3 vincristine), taxanes (6 paclitaxel, 2 docetaxel), 2 thalidomide, and 4 IFN-α. Two patients had also received radiotherapy. None of the patients had hepatitis B and C virus coinfection. All had normal clotting status at screening, and two had a history of thromboembolism. Although the protocol required a Karnofsky performance status >70%, an exception was granted to the patient with visceral involvement (Karnofsky index 60%) (Fig. 1).
cART, combined antiretroviral therapy; HHV-8, human herpesvirus type 8; KS, Kaposi's sarcoma; MDRD, Modification of Diet in Renal Disease; NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; pVL, plasma viral load.
Study treatment and efficacy
Two of the 12 enrolled patients discontinued the study, one on day 7 because of hives and one on day 13 because of acute renal failure in a context of chronic renal disease. The latter patient had gastrointestinal involvement and died at week 7 (Fig. 1). This patient had a high tenofovir plasma trough concentration at baseline (C24 h = 305 ng/ml). The remaining 10 patients were assessable at week 24. One patient (#4) had a lenalidomide dose reduction (15 mg/day) because of a rash during the first cycle, and this dose was maintained through week 48. Three patients (#2, #10, and #11) stopped the study treatment because of disease progression at week 16, 24, and 28, as evaluated by the study investigator and the patients themselves. Disease progression was confirmed by the ACTG score, with an increase of ≥25% in the size of the index lesions at week 24. All three patients had detectable HHV-8 viral load: patient #10 had, respectively, <10, 1,819, 7,133, and 709 copies/150,000 cells at week 0, 4, 12, and 24; patient #2 had <10, 28, <10, and <10 copies/150,000 cells; and patient #11 had 998, 15 365, 2,090, and 27 877 copies/150,000 cells. This last patient withdrew from the study on being diagnosed with multicentric Castleman's disease. This diagnosis was raised early after inclusion and confirmed during follow-up. Poor adherence was suspected in this case, as the HIV pVL was 54 copies/ml at week 4, the plasma ARV trough concentrations were below the LOQ at week 12, and lenalidomide pill consumption was below 90%.
At week 24, based on the investigator and patient assessments (Table 2), there was no PR or CR among the 10 assessable patients (95% confidence interval [CI]: 0–31). This was confirmed by the validation committee. Based on ACTG criteria, one patient (#3) had a PR at week 24 (10%, 95% CI: 0.2–44.5), with a 58% reduction in the total surface area of his index lesions. There were no additional responses at week 48 according to the investigator and patient assessments, while ACTG criteria showed three additional responses. Thus, a total of four patients had a PR at week 48 (40%; 95% CI: 12.2–73.8). The estimated KS progression-free survival rate at week 48 was 63.6% (95% CI: 35.4–84.8) (Fig. 2).
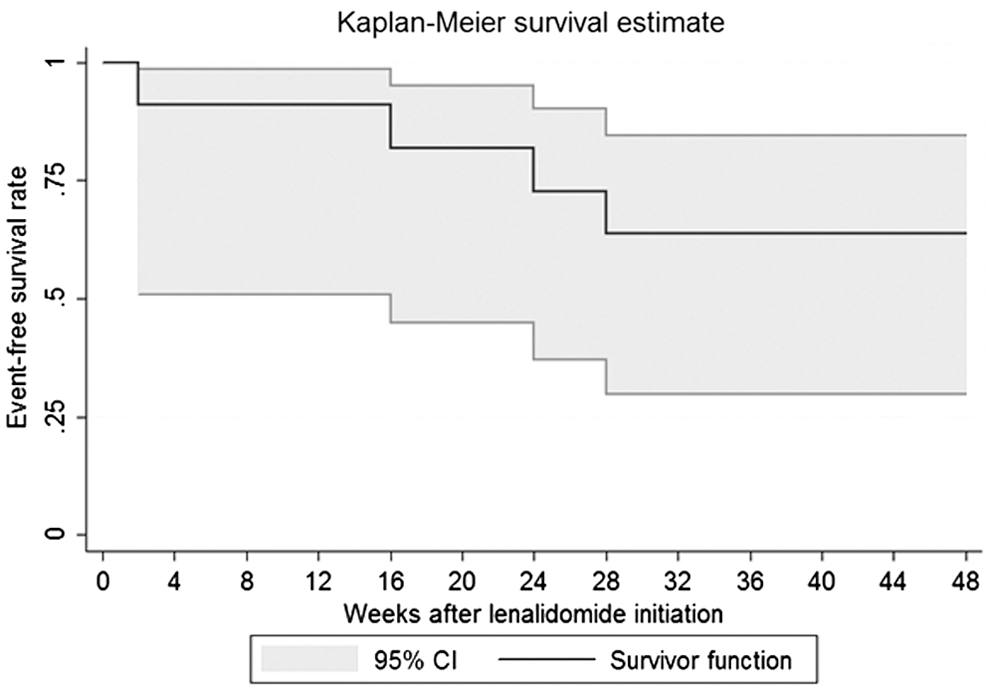
Kaplan–Meier curve of event-free survival in 12 patients treated with lenalidomide. The patient (#8) who experienced hives at week 1 was censored at this time. The four events correspond to a patient who died (#6) and three patients who experienced disease progression (#2, #10, and #11), as assessed by both the study investigator and the patient. The event-free survival rate at week 48 was 63.6% (95% CI: 35.4–84.8). CI, confidence interval.
Not assessable, patient left the study at week 24 for progression.
Not assessable, patient stopped the study treatment at week 16 for progression but was followed until week 48.
Not assessable, patient stopped the study treatment at week 28 for progression but was followed until week 48.
ACTG, AIDS Clinical Trials Group; PD, progressive disease; PGA, Physical Global Assessment; PR, partial response; SD, stable disease; SI, slight improvement.
Biological outcomes
During follow-up, two patients experienced an isolated increase in HIV pVL (54 copies/ml at week 4, returning to <50 copies/ml at week 8 with no change in cART, and 73 copies/ml at week 48). Changes in biological parameters between baseline and week 48 are described in Table 3. The only significant changes were a fall in leukocyte and neutrophil counts.
Bold, statistical significant values.
Safety
Lenalidomide was generally well tolerated. The main adverse events were neutropenia, thrombocytopenia, and asthenia (Table 4), as mentioned in the summary of product characteristics. During a total of 101 treatment cycles, seven patients experienced 14 grade 3–4 adverse events that the investigators considered at least possibly due to lenalidomide, of which six were classified as certainly due to lenalidomide. One patient died at week 7, as noted earlier.
Worst grade for each adverse event reported in the same patient and considered as the same occurrence is presented in this table.
Cytokine assays
Only IL-8 levels changed significantly between baseline and the end of lenalidomide treatment, with a median change of +0.90 (p = .028) (Table 5). There was no association between baseline cytokine plasma levels and the change in the total surface area of the KS index lesions at the end of lenalidomide treatment. By contrast, the decrease in IFN-γ and TNF-α plasma concentrations from baseline to the end of lenalidomide treatment was associated with a decrease in the total surface area of the index lesions (r = 0.818, p = .004 for IFN-γ, and r = 0.758, p = .011 for TNF-α). In addition, we observed a significant difference in the change in IFN-γ plasma concentrations (+37 vs. −1.6 pg/ml, p = .017) and IL-10 plasma concentrations (+19.6 vs. −0.1 pg/ml, p = .033) between the three patients with disease progression and the seven patients without disease progression (Table 5). No association was observed between the other cytokines and disease progression.
Bold, statistical significant values.
Nonparametric analysis of change in cytokine levels (Baseline − Last value), Wilcoxon signed-rank test.
Nonparametric analysis, Mann–Whitney test.
IFN, interferon; IL, interleukin; IQR, interquartile range; TNF, tumor necrosis factor, VEGF, vascular endothelial growth factor.
Drug plasma concentrations
ARV and lenalidomide trough plasma concentrations (C12 h or C24 h) were considered adequate during the entire study period. Lenalidomide pill consumption was generally high, as reported in Table 2, with six patients consuming more than 95% of lenalidomide pills. Two patients consumed between 90% and 95% of their pills in at least one period and the last two consumed between 86% and 90% in at least one period. We observed no lenalidomide accumulation in plasma, including in patients receiving a P-glycoprotein inhibitor such as ritonavir. One patient (#9) had a low darunavir C24 h at week 12 and 24 (1,164 and 455 ng/ml), probably owing to reduced intestinal absorption under possible fasting conditions, leading to transient HIV low-level viremia (73 copies/ml at week 48).
Discussion
We observed no improvement of KS during lenalidomide therapy, as evaluated by both the study investigators and the patients using the PGA score, while ACTG criteria suggested a partial improvement in 40% of patients at week 48. No CR occurred. The KS-event-free survival rate at week 48 was 63.6%.
Evaluation of treatment responses in cutaneous KS remains a challenge. The ACTG classification has been widely used since the pre-cART era. 8,33 However, this classification was validated in 199733 and new data suggest that, in the cART era, a refinement of the ACTG classification or another prognostic index could be of interest. 40,41 We chose a composite endpoint for our trial of lenalidomide as rated by the study investigators, the patients, and a validation committee. By comparison with the sensitivity of the ACTG criteria, we hoped that PGA, a more global evaluating test, could be better. This choice was made in 2009. PGA was used at this date as an acceptable evaluation criterion in psoriasis and cutaneous lymphoma for drug approval. 42 The ACTG criteria and the PGA are complementary. The PGA could have offered, in case of efficacy, a new method to evaluate cutaneous KS. The ACTG criteria, used as a secondary endpoint, showed a PR in one patient (10%) at week 24 and in four patients (40%) at week 48, corresponding to a ≥50% decrease in the total surface area of the index lesions. For future studies, the ACTG criteria must be used as primary criteria. Nevertheless, composite criteria such as the ACTG criteria and use of PET scan could be interesting. Previous reports of follow-up of KS therapy showed promising results with the positron-emission tomography (PET) in addition to conventional evaluation. 30,31,43 –45 PET showed a reduction in hypermetabolic abnormalities and confirmed PR or CR. This latter method has the advantage of being standardized and reproducible. Use of a clinical index combined with PET could be of interest in KS treatment evaluation and should be considered in future clinical trials. In our previous three case reports on the use of lenalidomide in KS in HIV-infected patients, we observed a successful effect and good tolerance after failure of HAART and several chemotherapies. 30
Lenalidomide was generally well tolerated in our study. Adverse events were comparable to those observed in 14 other clinical trials in hematologic disorders. 46 The most frequent adverse effect was myelosuppression with neutropenia, thrombocytopenia, or anemia (33%). No neurologic toxicity was observed. Adverse effects are reported to be more frequent with conventional KS therapy, the most common (>30%) being myelosuppression, neurologic toxicity, fever, cytolysis, rash, vomiting, and hypersensitivity reactions with IFN-α, bleomycin, vinblastine, doxorubicin, daunorubicin, docetaxel, and paclitaxel. 2,7,8
The overall response rate (ORR) observed in our trial based on the ACTG criteria was slightly smaller to that reported with conventional intravenous chemotherapies, for example, 61% with vincristine, 48% with bleomycin, and 59% with liposomal pegylated doxorubicin, 12,47,48 and similar to the ORR reported for immunomodulatory agents, bevacizumab (31%), 49 matrix metalloproteinase inhibitor COL-3 (29%–41%), 50 tyrosine kinase inhibitor imatinib (33%), 51 and thalidomide (34%–47%). 14,16 It is important to note that our patients had received multiple previous KS therapies. The median progression-free survival time of more than 12 months observed here with lenalidomide was longer than the 8.3 months reported elsewhere with bevacizumab, 49 5.2 months with imatinib, 51 and <4 months with historical drugs. Oral lenalidomide allows more convenient administration, avoiding overnight or 1-day hospitalization and improving patients' quality of life. 52 –54 Moreover, in contrast with many antineoplastic drugs, lenalidomide does not seem to interact with antiretroviral therapy as also observed in the present trial. 55 In a phase II trial of imatinib in HIV-KS, Koon at al. observed possible interactions with ARV, potentially resulting in added toxicity. 51 We observed no lenalidomide accumulation in plasma, even in patients receiving P-glycoprotein inhibitors such as ritonavir. These results are consistent with published data 56 –58 and support the use of lenalidomide in HIV-infected patients with malignancies such as multiple myeloma. Concomitant use of ARV and conventional chemotherapy can lead to life-threatening interactions with loss of efficacy and/or increased toxicity. 59 Our study focused only on refractory KS.
KS is a complex multifactorial disease due to an oncogenic virus, HHV-8, and is associated with chronic inflammation in a context of immunodepression. Lenalidomide targets multiple KS pathways and may thus be of interest in this disease. 8,13,60 Our study is the first to report plasma cytokine concentrations in KS-HIV patients treated with lenalidomide. We observed a substantial increase in the plasma IL-8 level from baseline, in keeping with reports in metastatic malignant melanoma. 61 The Il-8 increase may reflect T-cell activation induced by lenalidomide. In a paracrine manner, IL-8 may contribute to circulating monocytes and neutrophil recruitment into KS lesions. However, this increase may also contribute to angiogenesis leading to KS progression. Evolution of IL-8 may be reflected increasing T-cell activation and angiogenesis but with no influence of disease progression. The exact role of IL-8 could be explained in future studies. Despite the known impact of lenalidomide on proinflammatory cytokine production, we observed no fall in TNF-α, IL-1, IL-6, or IL-12 plasma levels. 62 Finally, we found a significantly larger increase in IL-10 and IFN-γ levels in patients with progressive disease than in those with stable or improved disease. These results are consistent with those obtained by Polizzotto et al. in multicentric Castleman's disease, suggesting the involvement of IL-10 in KS flares. 63
Pomalidomide is a new thalidomide analog approved in Europe and the United States in 2013 for hematologic diseases. 64 This drug shows better antitumor, immunomodulatory, and antiangiogenic properties and fewer adverse effects than thalidomide and lenalidomide. 29,64 Pomalidomide has also shown promising results in a phase I/II study, including both 15 HIV-infected patients and 7 uninfected persons, with 20% CR and 40% PR and a median time to response of 8 weeks in HIV-infected patients. 65 In view of the disappointing results obtained here with lenalidomide, pomalidomide should be considered for future trials in HIV-associated KS.
List of ANRS 154 Lenakap Trial Investigators
Dr. P. Chevojon, Prof. C. Lebbe, Dr. P. Genet, Dr. G. Pichancourt, Dr. A. Lafeuillade, Dr. P. de Truchis, Dr. L. Escaut, Prof. D. Salmon-Ceron, Dr. E. Mortier, Prof. J.M. Molina, Dr. J.M. Chennebault, Prof. P. Morlat, Prof. P. Yeni, Dr. C. Allard, Prof. F. Raffi, Dr. E. Lazaro, Prof. Michelet, Dr. M. Partisani, Prof. L. Bernard, Dr. M. Obadia, Prof. T. May, Dr. L. Cotte, Dr. L. Blum, and Dr. E. Aslangul.
Footnotes
Acknowledgments
This trial was sponsored and funded by ANRS and conducted with the support of Celgene. We acknowledge Prof. Eric Caumes, Olivier Chosidow, and Dr. Pascal Pugliese who reviewed all photographs as a validation committee member.
Author Disclosure Statement
No competing financial interests exist.