
Abstract
Angola has an extremely diverse HIV-1 epidemic fueled in part by the frequent interchange of people with the Democratic Republic of Congo (DRC) and Republic of Congo (RC). Characterization of HIV-1 strains circulating in Angola should help to better understand the origin of HIV-1 subtypes and recombinant forms and their transmission dynamics. In this study we characterize the first near full-length HIV-1 genomic sequences from HIV-1 infected individuals from Angola. Samples were obtained in 1993 from three HIV-1 infected patients living in Cabinda, Angola. Near full-length genomic sequences were obtained from virus isolates. Maximum likelihood phylogenetic tree inference and analyses of potential recombination patterns were performed to evaluate the sequence classifications and origins. Phylogenetic and recombination analyses revealed that one virus was a pure subtype J, another mostly subtype J with a small uncertain region, and the final virus was classified as a H/U/CRF02_AG recombinant. Consistent with their epidemiological data, the subtype J sequences were more closely related to each other than to other J sequences previously published. Based on the env gene, taxa from Angola occur throughout the global subtype J phylogeny. HIV-1 subtypes J and H are present in Angola at low levels since at least 1993. Low transmission efficiency and/or high recombination potential may explain their limited epidemic success in Angola and worldwide. The high diversity of rare subtypes in Angola suggests that Angola was part of the early establishment of the HIV-1 pandemic.
A
In sub-Saharan Africa almost all subtypes, CRFs, and URFs circulate; the highest genetic diversity has been observed in West Central Africa where the HIV-1 pandemic is believed to have originated. 5 –7 On a global perspective, the most prevalent HIV-1 subtypes are C (50%), A (12%), B (11%), followed by CRF02_AG (8%), G (5%), CRF01_AE (5%), and D (2%). The remaining subtypes and recombinant strains represent less than 1% of HIV-1 infections. 5,6,8 Currently there are only three full-length genomes of subtype J and four of subtype H available in the Los Alamos HIV sequence database. 4 Full-length subtype J genomes are from Sweden and Cameroon (GenBank accession numbers AF082394, AF082395, GU237072) whereas subtype H genomes are from Belgium, United Kingdom, and Central African Republic (AF190127, AF190128, FJ711703, AF005496). In addition, subtype J appears as fragments in several CRFs and shorter sequences. 4
Angola is a South-western African country surrounded by Namibia, Zambia, Democratic Republic of Congo (DRC), and Republic of Congo (RC). According to the UNAIDS report, HIV/AIDS prevalence among adults in Angola was 2.4% in 2014. 9 Despite this low prevalence, all subtypes except B and many CRFs and URFs have been detected in Angola, in particular in the Provinces of Luanda and Cabinda where most studies have been done. This high diversity is a direct consequence of the long-standing presence of HIV-1 in the country and the frequent interchange of people with the DRC and RC. 10 –14 In this study we describe the first near full-length HIV-1 genomic sequences from HIV-1 infected individuals from Cabinda, a province of Angola, which is an exclave surrounded by DRC in the south and east, and by the RC in the north.
Blood samples from HIV-1 infected individuals were collected in 1993 from Hospital Distrital de Cabinda, Cabinda, Angola. Samples were collected anonymously with oral consent. The study was approved by the ethics committees of the participating institutions. Virus isolates were obtained using the co-cultivation method as described previously.
15
All three isolates used the CCR5 co-receptor.
16
Viral genomic RNA was extracted from cell culture supernatant and reverse transcriptase–polymerase chain reaction (RT-PCR) was performed using Titan One Tube RT-PCR System (Roche Diagnostic Systems). All amplifications were performed using the Expand Long Template PCR system (Roche Diagnostic Systems) according to the manufacturer's instructions. New primers were designed to amplify the full-length genomes (Supplementary Table S1; Supplementary Data are available online at
An overall genome-wide tree analysis of 93AOHDC250 and 93AOHDC253 showed that they clustered with subtype J reference sequences and were more closely related to each other than to the other J isolates, which is consistent with their epidemiological data (Fig. 1). Note that this tree is not a true phylogeny as it cannot depict the evolutionary history of all recombinant sequences; we used it here merely to investigate the overall sequence-based similarity for our classification purpose.
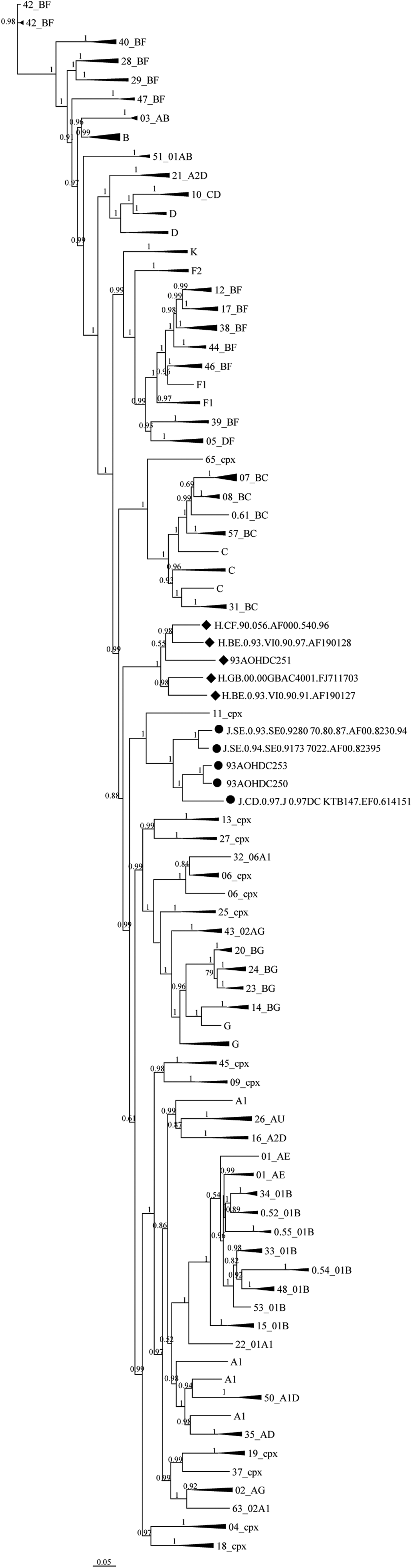
Overall tree classification of HIV-1 near full-length genomes of isolates 93AOHDC250, 93AOHDC251, and 93AOHDC253. ML trees were constructed with our sequences (highlighted by dots) and 136 reference sequences representative of all HIV-1 group M subtypes. Cladistics support is indicated by aLRT values. The scale is in units of substitutions per site. aLRT, approximate likelihood-ratio test; ML, maximum likelihood.
Bootscanning analysis showed no evidence of recombination in isolate 93AOHDC253 (Fig. 2A). Isolate 93AOHDC250 displayed an untypable region between positions 1450 and 2050 corresponding to the end of gag gene, the protease (PR) region, and the first 192 nucleotides of RT, which was further confirmed by phylogenetic analysis (Fig. 2B). BI results confirmed that 93AOHDC253 was a pure subtype J and that 93AOHDC250 had a genomic region with no known subtype in this gag/pol region (Fig. 3).

Genomic segment analysis of near full-length genomes. Panels show bootscanning and tree analyses of isolates 93AOHDC253
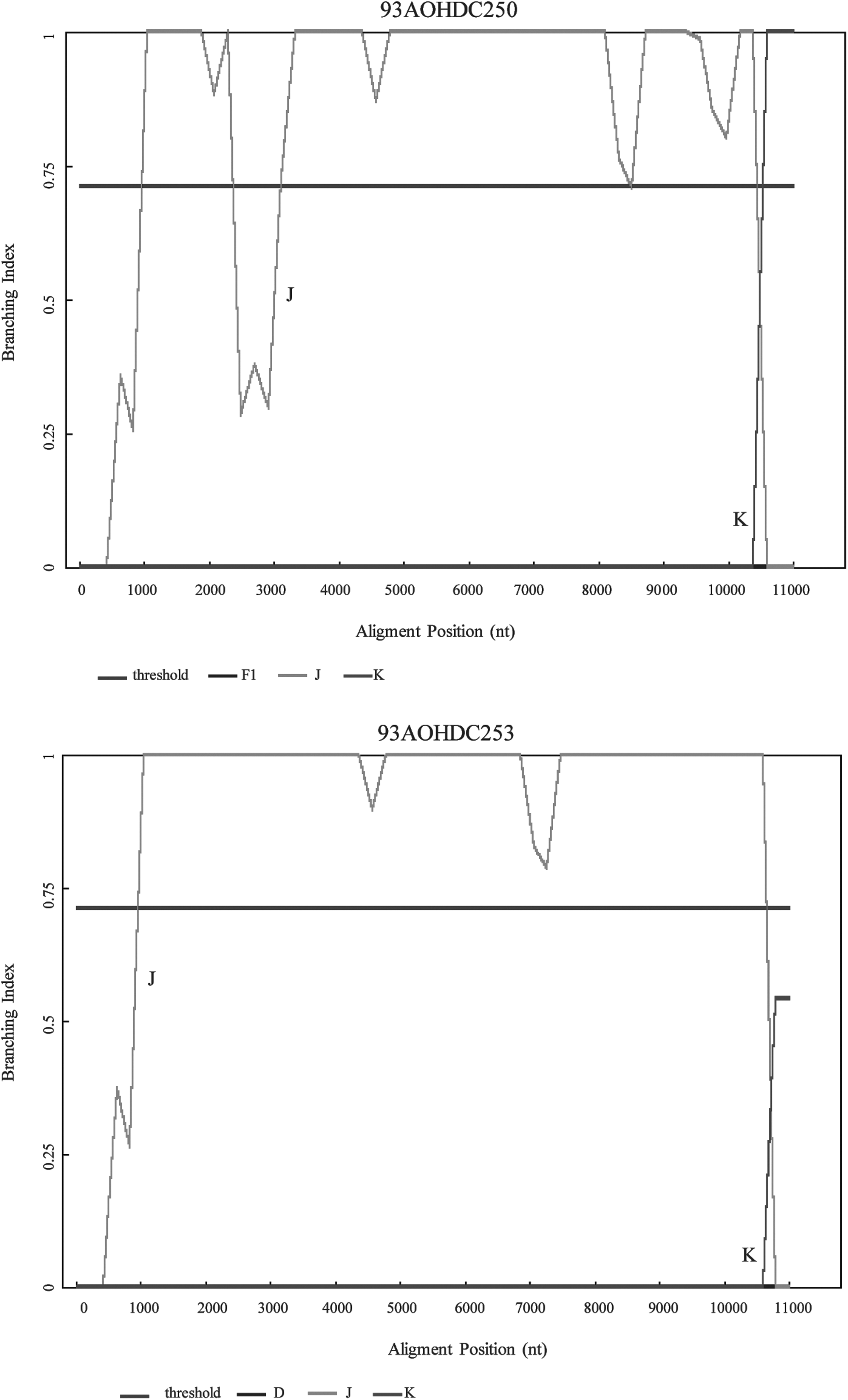
Branching Index (BI) analysis of 93AOHDC250 and 93AOHDC253.
Comparing our new J sequences to existing J sequences in the Los Alamos HIV database revealed that subtype J is quite diverse (Fig. 4). In the region where most J sequence fragments exist (HXB2 positions 7041–7358), 93AOHDC250 and 93AOHDC253 cluster together with a previous sequence from Angola, 93AOHDC247. 10 Interestingly, Angolan J sequences occur throughout the phylogeny of this env region, suggesting that the J epidemic in Angola is either the origin of subtype J or that there has been a lot of influx of subtype J from other geographic regions. The great diversity of subtype J has been previously noted in analyses of J-containing CRF11 and CRF13 genomes. 22,23
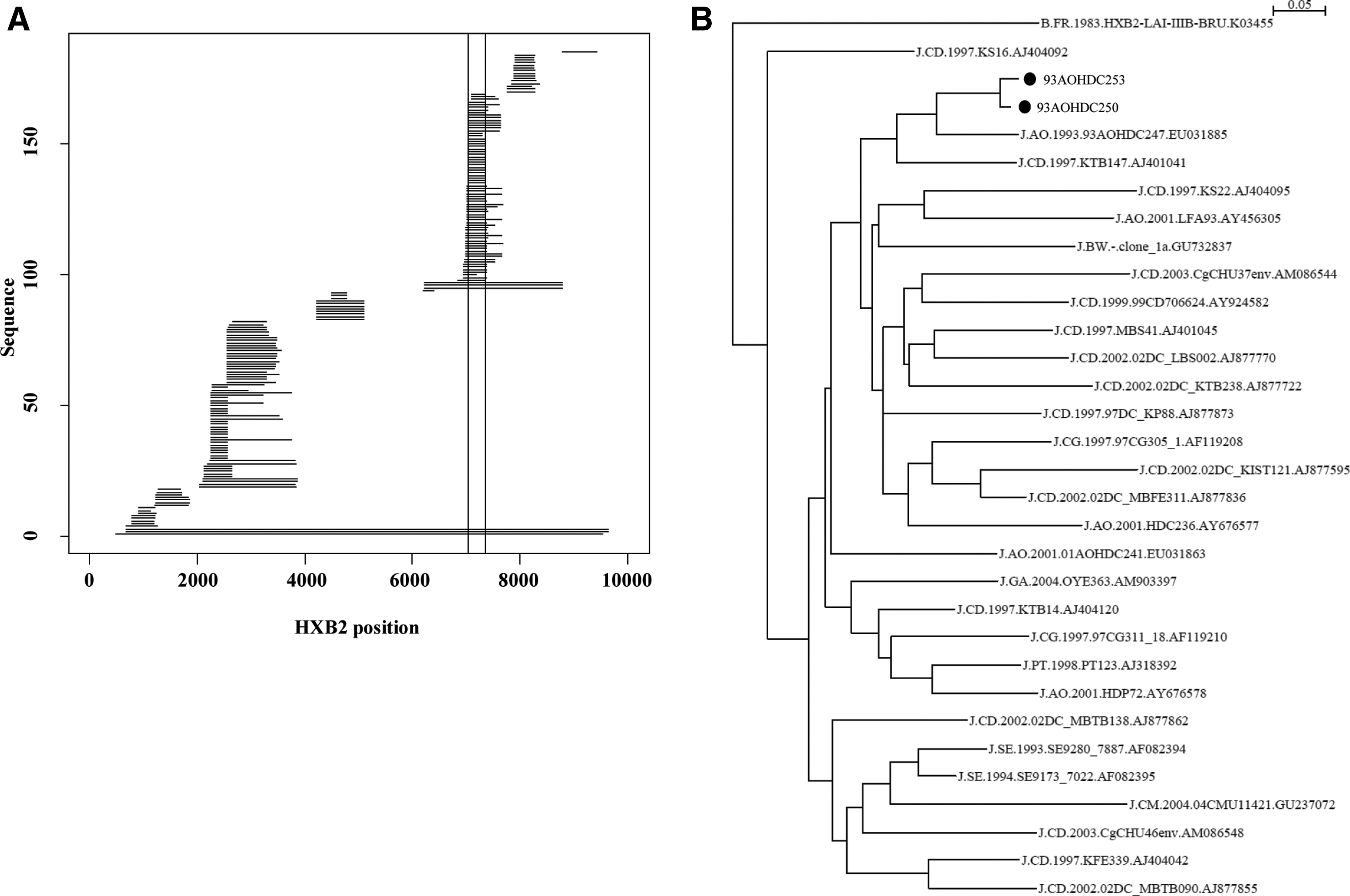
Subtype J env fragment comparison. At the time this report was written 185 subtype J sequence fragments were available in the LANL HIV database. Their HXB2 coordinates covered various parts of the HIV-1 genome
The overall tree analysis showed that 93AOHDC251 clustered with subtype H reference sequences (Fig. 1). Bootscanning analysis indicated an unclassifiable (U) region between positions 4936 and 5167 (corresponding to vpr gene), and a region between positions 8151 and 8888 (corresponding to the nef gene and 3′LTR) appeared to cluster between subtypes A1 and G (Fig. 2C). BI analyses again confirmed these results, and suggested that the nef/LTR region was below the subtype-defining threshold for subtype G (Fig. 3). Upon closer inspection using RIP, comparing this region to subtypes A1, G, and CRF02, it became clear that this region was in fact derived from CRF02; 93AOHDC251 was closer to CRF02 than either A1 or G in all parts of this region, which included a CRF02 A/G breakpoint (Fig. 5). Together, these analyses led to a classification of this genome sequence as the first H/U/CRF02_AG recombinant.

RIP analysis of segment III of 93AOHDC251. A sliding window analysis (window size 200 nt, step 1 nt; significance threshold 0.90) comparing 93AOHDC251 to consensus subtype sequences A1 and G, and CRF02 prototype sequence IBNG showed that this region was closer to CRF02 in both the A1 and G parts. The CRF02 A1/G breakpoint is indicated by a bashed vertical line. The top bar indicates statistical significance, and the lower bar best match.
In this report we describe the first three HIV-1 genomic sequences from Angola, a country that with the DRC and RC had a crucial role in the early dissemination of the HIV-1 epidemic. 7,10 Sequences were derived from isolates obtained in 1993 from patients living in Cabinda, a province in the North that borders both DRC and RC.
One isolate was a pure subtype J, another a J with an at this point unclear segment, and one was an H-based recombinant. The unclear region in 93AOHDC250 may be a divergent J segment or a segment of an as yet undiscovered subtype. As mentioned above, subtype J has previously been described as very diverse and may hide further sub-subtypes or recombinants of sub-subtype nature. 22
Despite being in circulation in Cabinda since 1993, as of February 2015 only 27 J and 73 H sequences from Angola had been deposited in the Los Alamos HIV Sequence Database (3.1% and 8.3% of respective subtype sequences from Angola). Globally, pure subtypes J and H are also very rare, which suggests low biological fitness, low transmissibility, high recombination potential, and unsuccessful introductions into high-risk populations. However, the isolates described herein replicate to normal levels in different cell types suggesting that their biological fitness is no different from other subtypes. 16 Interestingly, we have found that our J and H isolates are much more sensitive (about nine times) to the CCR5-antagonist TAK-779 when compared with isolates from other subtypes that are more prevalent in Angola and Portugal. 16 This suggests poor binding to the main HIV co-receptor (CCR5), which may indicate transmissibility problems of these subtypes. On the other hand, J and H subtypes have been found in many recombinants such as CRF04_cpx, CRF06_cpx, CRF11_cpx, CRF13_cpx, CRF18_cpx, CRF27_cpx, and CRF49_cpx. 17 Thus, the rarity of pure subtype J and H, together with the large HIV-1 diversity in Angola and neighboring countries, which suggests they have been around for long times, suggests that these subtypes are less fit to transmit or establish infection by themselves. Similar to how latent virus may survive within a host through recombination with nonlatent immune escaping plasma virus, subtype J and H virus may do better if they recombine with more fit subtypes. 24
HIV-1 subtypes J and H are present in Angola at low levels since at least 1993. The high diversity among Angolan subtype J env sequences and the fact that the rare subtype H has recombined in Angola together suggest that Angola is either the origin of subtype J or, more complicated, that there has been a lot of influx of subtype J from other geographic regions. Low transmission efficiency and/or high recombination potential may explain their limited epidemic success in Angola and worldwide.
Footnotes
Acknowledgments
Financial support for this research was provided by the Fundação para a Ciência e a Tecnologia (FCT), Portugal (project PTDC/SAU-EPI/122400/2010), part of the EDCTP2 program supported by the European Union. Rita Calado is supported by Fundação para a Ciência e a Tecnologia (FCT), Portugal (grant no. SFRH/BD/70715/2010). I.B. was supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal (grant no. SFRH/BPD/76225/2011). Thomas Leitner was supported by the National Institutes of Health (NIH), USA (grant no. R01AI087520).
Sequence Data
Sequences have been assigned with GenBank accession numbers KU310618, KU310619, and KU310620.
Authors' Contributions
Conceived and designed the experiments: I.B., T.L., and N.T. Performed the experiments: I.B., P.B., T.L., and R.C. Analyzed the data: I.B., T.L., R.C., and N.T. Wrote the article: I.B., R.C., T.L., and N.T. The final text was read and approved for submission by all authors.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.