
Abstract
Any method for silencing the activity of the HIV-1 retrovirus should tackle the extremely high variability of HIV-1 sequences and mutational escape. We studied sequence variability in the vicinity of selected RNA interference (RNAi) targets from isolates of HIV-1 subtype A in Russia, and we propose that using artificial RNAi is a potential alternative to traditional antiretroviral therapy. We prove that using multiple RNAi targets overcomes the variability in HIV-1 isolates. The optimal number of targets critically depends on the conservation of the target sequences. The total number of targets that are conserved with a probability of 0.7–0.8 should exceed at least 2. Combining deep sequencing and multitarget RNAi may provide an efficient approach to cure HIV/AIDS.
Introduction
R
Materials and Methods
Collection of samples and virus isolation
RNA preparations were provided by the State Research Center of Virology and Biotechnology Vector (Russia) from their collection of isolates. Five isolates of HIV-1 subtype A (10RU6587, 11RU6933, 11RU6949, 10RU6483, and 11RU1996), which were collected by N.M. Gashnikova, were used. RNA was extracted from 500 μL of plasma samples using ViroSeq reagents (Celera Diagnostics), and then treated with DNase using a Turbo DNA-free kit (Ambion). The concentration of RNA preparations was measured using a NanoDrop 2000.
Reverse transcription polymerase chain reaction (RT-PCR)
Five RNA preparations from five isolates were pooled (6 ng of each) and used for RT-PCR. Approximately 15 ng of total RNA and M-MLV reverse transcriptase were used to synthesize cDNAs by the use of a Turbo DNA-free kit (Ambion) according to the manufacturer's instructions. cDNAs corresponding to five regions in the viral RNA were synthesized using the primers indicated in Table 1. Nested PCR was used for amplification of regions of ∼300 bp that contained the selected RNAi targets.
6
Primers were selected using the Primer Selection Tool (
Sequences 1–20 or 21–25 were selected using the corresponding regions in AF316544 or AF286250, respectively.
Luciferase reporter assays
HEK293T cells were plated 1 day before transfection (5 × 105 cells per 60-mm culture dish). To prepare liposomes, 10 ng of the experimental DNA constructs containing the targets of RNAi cloned into the psiCHECK-2 (Promega) vector were mixed with 100 ng of the corresponding DNA constructs that expressed the RNA hairpins cloned into the GeneClipU1 Neomycin vector (Promega) in 600 μL of serum-free medium. Next, 1 μL of TransFast reagent (Promega), diluted according to the manufacturer's recommendations, was added, and the mixture was incubated at room temperature for 15 min. The cell suspension (0.2 mL, ∼5 × 104 cells) was centrifuged at 2,000 rpm in 1.5-mL Eppendorf tubes for 3 min at room temperature, and the precipitate was mixed with 60 μL of the liposome-containing sample. After incubation for 1 h at 37°C, the transfected cells were transferred into a 24-well plate containing 0.5 mL per well of the cell culture medium supplemented with serum, and cells were incubated for 72 h. Firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) and a Reporter Microplate Luminometer (Turner BioSystems). The Renilla luciferase data were normalized to the firefly luciferase data. Excel and Origin software were used for data analysis.
Results
Our aim was to determine the variability in RNAi targets to develop an optimal scheme of RNAi for silencing the activity of HIV-1 and to circumvent the problems associated with the high variability of HIV-1 sequences. In this article, the set of sequences obtained for five Russian isolates is treated as a model of an individual patient. Such a set can mimic the variability of RNAi targets for multistrain HIV-1 patients and/or the development of multiple strains over time for a particular patient. As we are primarily interested in assessing the characteristic quantitative variability of RNAi targets, such an approach seems reasonable. To ensure reliable statistics for the rather small set of data, the study of variability was extended to 300 nt fragments that included five 23 nt targets. Positions of the targets are shown in Figure 1 and designated as A1, A2, A3, A4, and A6.
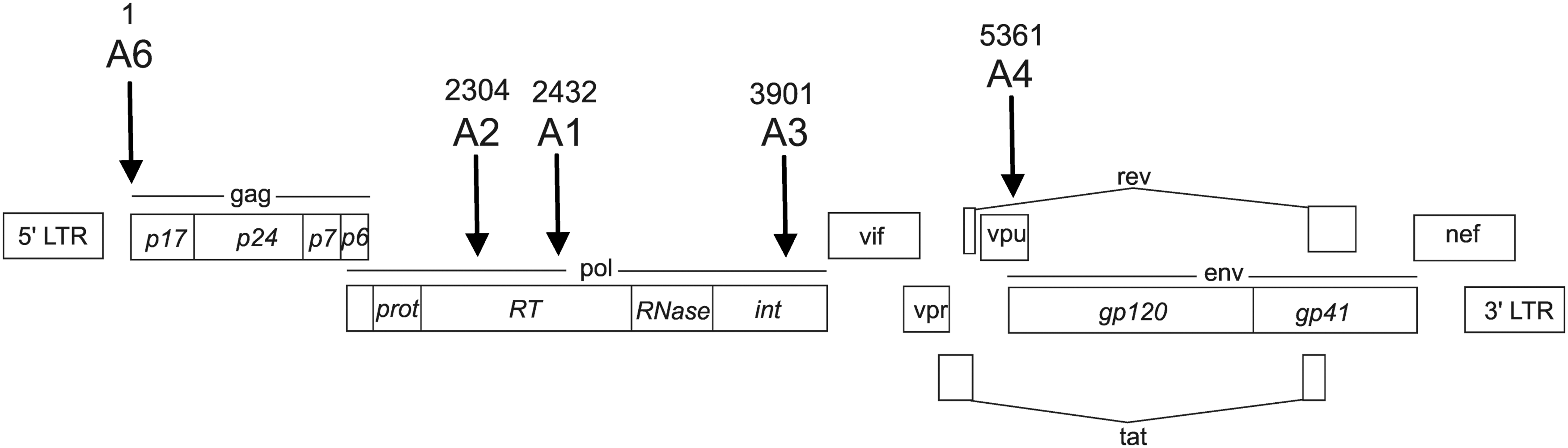
Schematic presentation of RNAi targets (A1–A5, and A6) within the HIV-1 gene map. The 23-bp targets are located in the sequenced regions of about 300 bp in length in HIV-1 subtype A isolates in Russia. The values shown above the targets indicate the 5′ numbering of a target in the reference sequence (GenBank AF316544). A1–A3 are located inside the pol domain, A4 inside vpu, and A6 inside the gag domain. The corresponding 300 bp sequences comprising the A1–A5 and A6 RNAi targets are shown in Supplementary Fig. S1.
Variations in the vicinity of RNAi targets
As a convenient reference, we added to our data set the corresponding fragments from the genome sequence for HIV-1 isolate 97CDKP58e from the Republic of the Congo (GenBank Accession No. AF316544; hereafter HIV-1 Congo). The sequence for HIV-1 Congo is commonly considered as a generic reference for HIV-1 subtype A sequences. The general (dis)similarity between 300 nt fragments around RNAi targets for the mixture of five Russian isolates and HIV-1 Congo is shown in Figure 2 and is characterized in terms of maximum-likelihood trees. The trees were obtained by the tools based on the PhyML algorithm
7
that are built into the UGENE package [8]. The distances on the branches reveal that the 300-nt fragments for HIV-1 Congo are clearly more distant from the counterpart fragments for the five Russian isolates, which are rather close to each other. The details of genetic variations (mutations, stop codons, and microindels) responsible for the divergence between the 300-nt fragments around the RNAi targets can be found in the Supplementary Fig. S1 (Supplementary Data are available online at
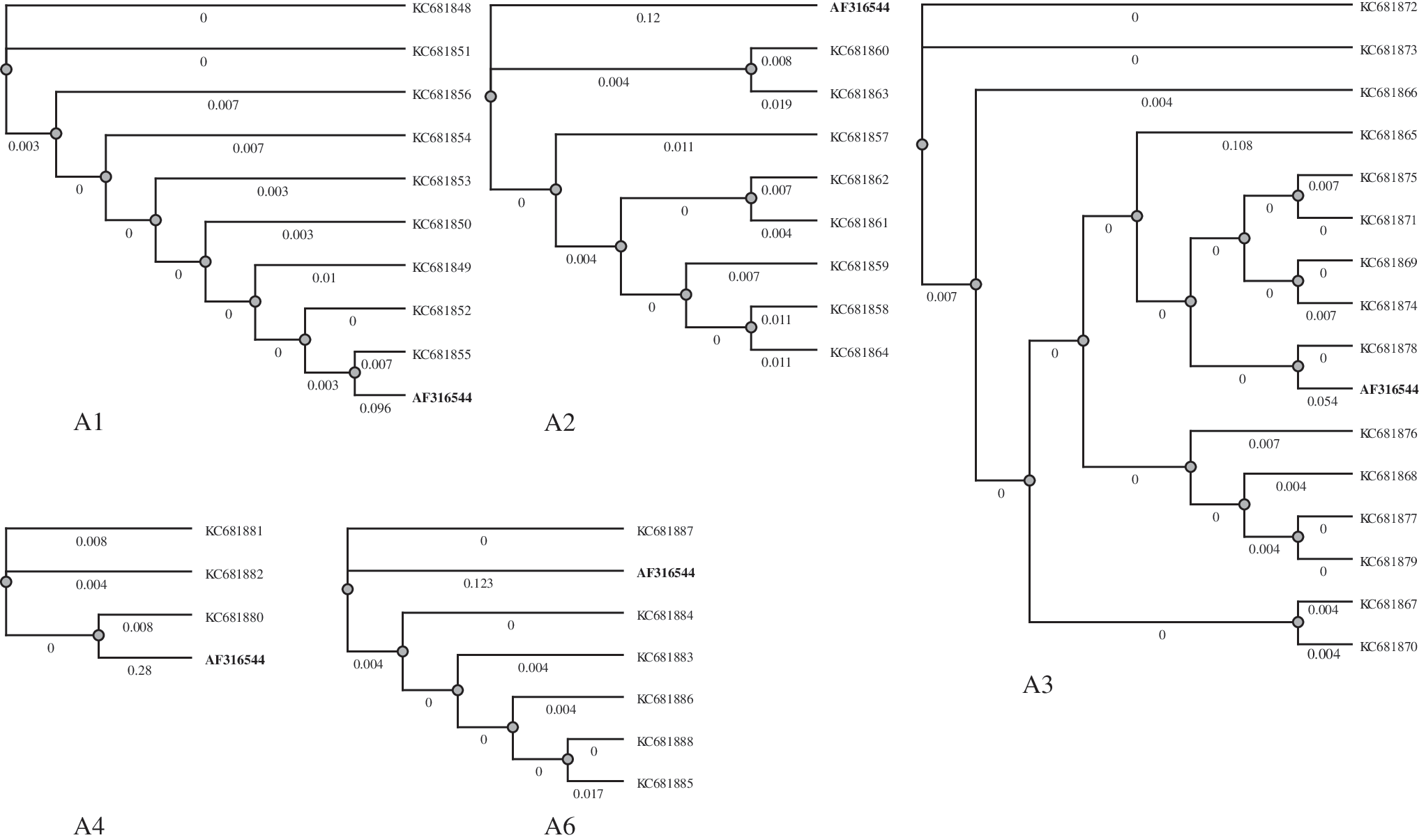
Maximum-likelihood trees for the 300-nt sequences comprising RNAi targets for the mixture of five Russian HIV-1 subtype A isolates. The corresponding sequences for HIV-1 isolate 97CDKP58e from the Republic of the Congo (AF316544) are boldfaced. A1–A6 refer to RNAi targets.
The mutations in the sequenced 300-nt fragments around RNAi targets were counted relative to a chosen reference sequence. Preliminarily, all sequences were aligned using the UGENE toolkit.
8
The frequency of mutations was assessed by two methods. In the first method, the corresponding fragments in the genome sequence for HIV-1 Congo were used as the relevant reference sequences (Supplementary Fig. S1). The frequency of mutations fm
is defined in this case as
where Nm
is the complete number of mutations detected in the set of 300-nt fragments corresponding to a particular RNAi target, Ns
is the number of 300 nt fragments, and Lref
is the length of the reference sequence. In the second method, we used jackknife (leave-one-out) resampling
9
in the set of sequenced 300 nt fragments corresponding to a particular RNAi target. In this procedure, one of the 300-nt fragments is picked as the reference sequence. The mutations are counted in the remaining
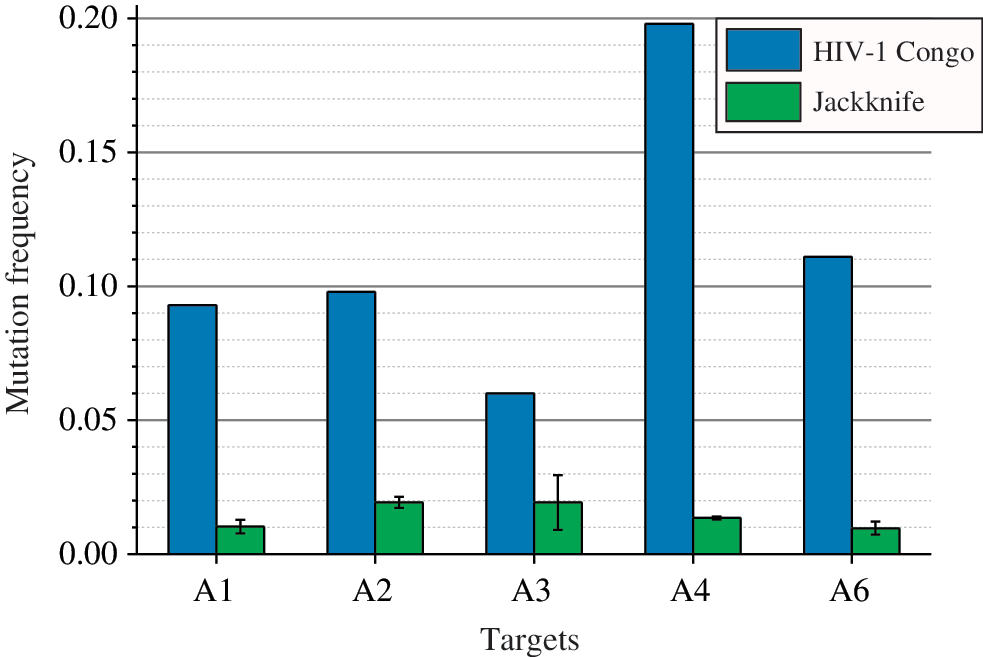
The mean frequencies of mutations in the 300-nt sequences comprising RNAi targets for the mixture of five Russian HIV-1 subtype A isolates. The frequencies were assessed either relative to the corresponding fragments of HIV-1 isolate 97CDKP58e from the Republic of the Congo (AF316544) or by jackknife resampling of the sets for five Russian isolates, as described in the main text. All sequences were preliminarily aligned using the UGENE toolkit [8]. Color images available online at
In addition to variations related to point mutations, we also detected a few microindels in the vicinity of RNAi targets. These rare events cannot be used for statistical assessment of corresponding frequencies and provide only rough estimates, that is, 10−3–10−4 (in the first method) and 10−4–10−5 (in the second method). The variations related to microindels are two to three orders of magnitude less frequent than that of mutations and are not essential for subsequent estimates.
Mutations in RNAi targets
The frequencies of mutations determined in the vicinity of RNAi targets can be extrapolated to assess the probability of mutations directly in the targets. Such an extrapolation is much more statistically reliable than direct counting of mutations in the targets for the given set of data. The latter procedure would need data that were several tens of times more extensive.
Assuming Poisson statistics, the probability of detecting k mutations in a target of length Lt
is given as
where fm corresponds to the frequency of mutations assessed for the set of 300 nt fragments around a particular target. The conservation of the RNAi target corresponds to the absence of mutations, k = 0. The length of RNAi targets was Lt = 23 nt. The results of these straightforward computations are summarized in Figure 4.
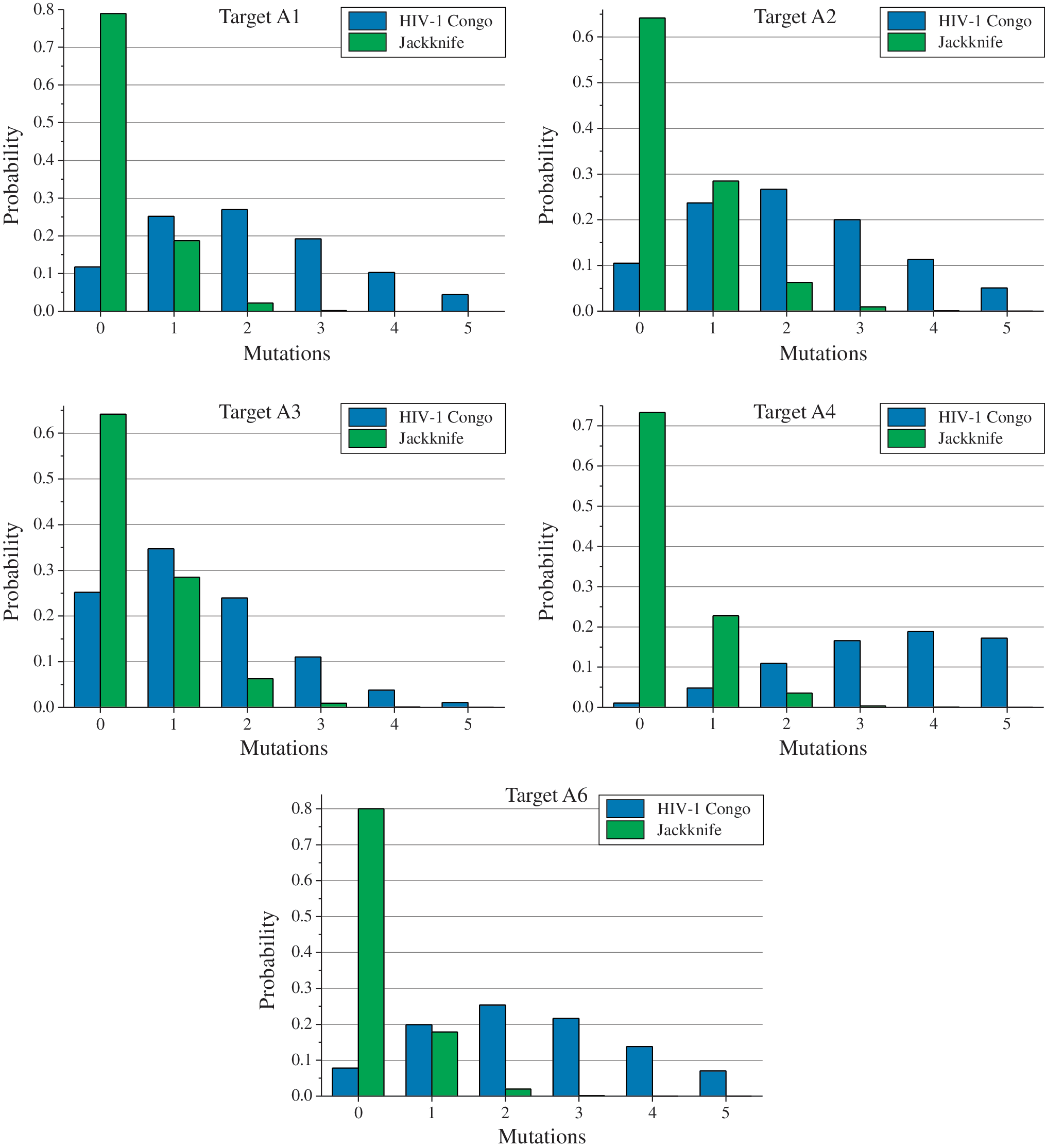
The probabilities of detecting a fixed number of mutations in the different RNAi targets. The probabilities were calculated by applying Poisson statistics [Eq. (2)] to the targets with a length of 23 nt. The mean frequencies of mutations in the RNAi targets were assessed by the number of mutations in the 300-nt sequences comprising RNAi targets for the mixture of five Russian HIV-1 subtype A isolates (Fig. 3). The frequencies were assessed either relative to the corresponding fragments for HIV-1 isolate 97CDKP58e from the Republic of the Congo (AF316544) or by jackknife resampling of the sets for five Russian isolates, as described in the main text. All sequences were preliminarily aligned using the UGENE toolkit [8]. Color images available online at
Multitarget RNAi
Now we are able to illustrate how the determined probabilities of detecting mutations in RNAi targets (Fig. 4) can be used to choose the optimal treatment for HIV-1 patients. Although the sequence for HIV-1 Congo may be efficiently applied to the assessment of evolutionary divergence between HIV-1 sequences, the corresponding fragments cannot be used as a standard for RNAi targets. RNAi needs the complete correspondence in RNA duplexes, whereas none of the target fragments for HIV-1 Congo remains sufficiently conserved (Figs. 3 and 4).
Consider the reciprocal situation. Let the set of RNAi targets for a particular patient be determined by deep sequencing. Our estimates indicate that these targets will be conserved with a probability in the range of 0.7–0.8 (Fig. 4). The probability
For Poisson distribution, the corresponding probabilities are given by
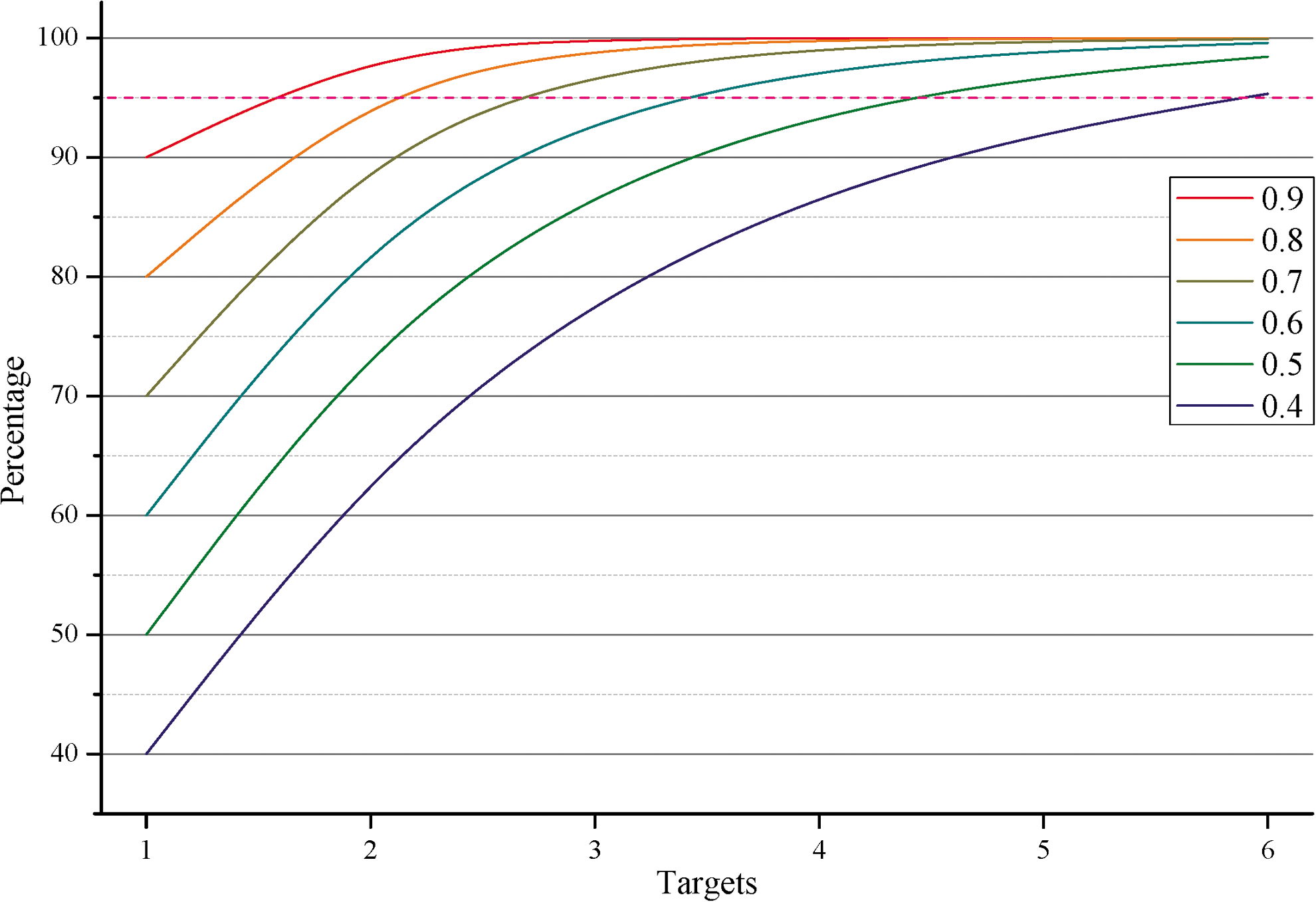
The dependence of the percentage of patients in which HIV-1 activity can be silenced by RNAi on the number of the RNAi targets with different levels of conservation. The efficiency and conservation of each target are implied to be approximately the same for the particular probability of conservation. The percentage dependencies were calculated by Eq. (3). The horizontal broken line indicates the threshold of 95% confidence interval recommended by the World Health Organization. Color images available online at
To test this conclusion experimentally, we used the luciferase reporter assay and cloned two targets corresponding to the A1 and A6 regions into the psiCHECK-2 vector (see the corresponding sequences in Supplementary Fig. S1). The vector was specifically designed for quantitative measurement of RNAi. XhoI and NotI cloning sites within the 3′ UTR of the Renilla luciferase gene were used for cloning of both targets as shown in Figure 6A. Construct 1 possessed an A3 target that perfectly corresponded to the shRNA and an A6 target that contained one mismatch. In contrast, Construct 2 possessed an A3 target with a single mismatch and an A6 target that perfectly matched the corresponding shRNA. Both shRNAs were expressed by the GeneClipU1 plasmid. In cotransfection experiments, we used a mixture of Constructs 1 and 2 (10 ng each) and the plasmids expressing the shRNAs. The data shown in Figure 6B indicate that, separately, the A3 or A6 siRNA had little effect since a single construct in the mixture could be strongly attacked by the siRNA that perfectly matched the target. In contrast, when both siRNAs were expressed, the efficiency of the RNAi was dramatically increased. The data strongly support our conclusion that multiplicity of RNAi targets could overcome their variability.
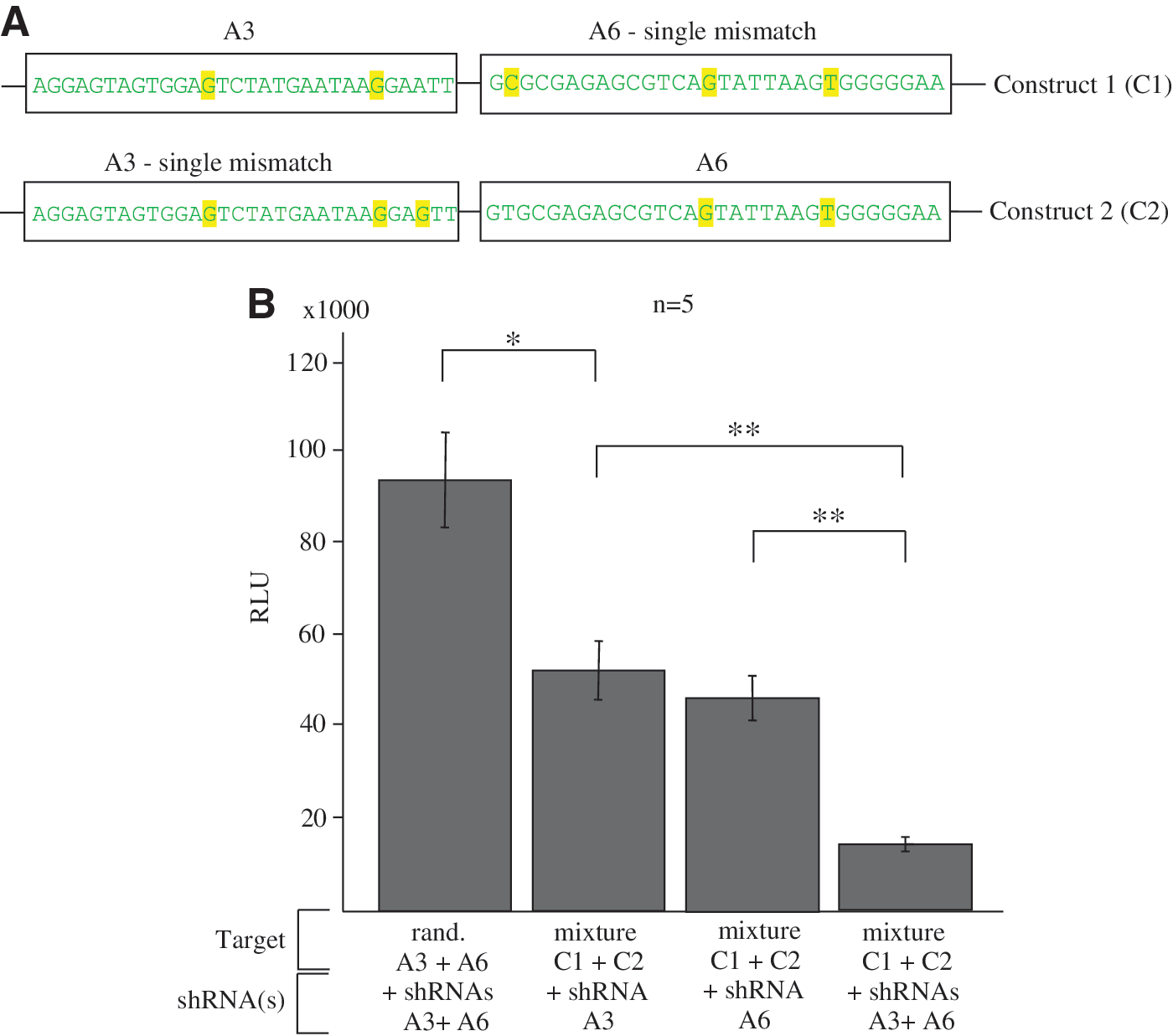
Analysis of the efficiency of RNAi initiated by the transfected plasmids expressing shRNAs and the mixture (1:1) of two constructs.
Discussion
The HIV-1 retrovirus has acquired a number of molecular mechanisms that allow escape from the protective immunity of the host, as well as current antiretroviral therapy. One of the mechanisms is the high variability of the viral genome, which is mainly due to the error-prone nature of the viral reverse transcriptase. 10 Ironically, the host-protective mechanism that induces G-to-A substitutions in the viral RNA by deoxycytidine deaminase APOBEC3G—which is able to suppress replication of different viruses possessing a single-stranded DNA intermediate by inducing mutagenesis and functional inactivation of the virus—is also a major source of HIV-1 variability. 11,12 One more source of HIV-1 variability is the recombination-prone nature of the viral reverse transcription machinery that leads to unusually frequent associations between distinct viral genotypes. 13,14
Our analysis of mutation frequencies in different codon positions in the targets A1–A3 in HIV-1 subtype A isolates in Russia suggests that the most conservative positions are the first and second positions. 15 The data indicate that there is a positive selection for biologically active viruses in the course of infection.
Whatever mechanism is the cause of frequently occurring mutagenesis in the HIV-1 genome, the monitoring of the sequences of selected RNAi targets should be the first step in the development of RNAi-based gene therapy of HIV/AIDS. Genetic constructs expressing a single or several perfectly matched 21-nt siRNAs in transfected cells lead to efficient attack of the corresponding target in HIV-1. 16 –19 Although a new approach of using the CRISPR gene editing technique was suggested for the removal of HIV from the DNA of human cells, 20 we believe that RNAi-based technology is closer to practical use. This antiviral technology has a number of advantages over traditional antiviral therapy. It uses an etiological approach as well as highly potent antiviral siRNA molecules as an agent with preventive and therapeutic value. 3 siRNAs can be designed and synthesized much more easily than traditional antiretroviral drugs. Moreover, a particular siRNA can be used in combination with other siRNAs to reduce the possibility of imperfect correspondence between a single siRNA and its target in the mixture of viruses in a patient.
Multiplying noisy or unreliable elements to improve the functioning of a whole system is a common strategy both in engineering and in living organisms. The greater the variability in the RNAi targets, the larger their number should be. Our estimates indicate that RNAi for silencing of HIV-1 needs at least two targets. Although a fragment can work as an RNAi target only under a set of strict conditions, 21 the search for two or three targets in the whole genome of HIV-1 seems to not be especially limiting. Duplexes with one or two mismatches cannot participate efficiently in RNAi but are thermodynamically stable and may also partially silence the activity of HIV-1.
The repertoire of RNAi target sequences related to particular subtypes of HIV-1 and/or a particular population should be determined through data stored in available databases 22,23 or through specific projects. As a medical cure, the RNA duplexes corresponding to the targets for the particular patient can be picked from a pre-synthesized pool of targets that are typical of a particular population. The sequences for targets corresponding to a particular patient should be preliminarily determined before treatment by either deep sequencing or by using custom oligonucleotide microarrays. 24 –26 We believe that combining deep sequencing and multitarget RNAi may provide an efficient technique to cure HIV/AIDS.
Footnotes
Acknowledgments
We thank Y.N. Toropchina for technical assistance and Dr. M.P. Gashnikova for the help in some experiments. This work was supported by a grant from the Russian Science Foundation (project no. 15-14-00005).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.