
Abstract
Historically, coinfection of HIV and hepatitis C virus (HCV) was frequent among Chinese former blood donors (FBDs). This is largely due to ignorance/lack of education regarding appropriate sterilizing techniques and/or the availability of single-use needles and equipment. Although HCV shares identical transmission routes with HIV, the source of HCV in the Chinese blood donor population still remains unknown. In this study, we investigated the evolution and transmission of HCV and HIV in the Chinese FBD group. Similar to previous reports, two HCV subtypes (HCV 1b and 2a) and one HIV subtype (Thai-B) were identified in FBDs. The HCV 1b subtype had a similar evolutionary rate of 1.9 × 10−3 substitutions/site/year to that of HIV (2.06 × 10−3 substitutions/site/year), while the HCV 2a subtype had a faster evolutionary rate of 3.8 × 10−3 substitutions/site/year. Phylogeographical analysis indicated that the introduction of HCV 1b into FBDs was estimated to be earlier than that of HCV 2a and HIV (late 1970s vs. late 1980s). Bayesian Skyline Plot (BSP) analysis further confirmed our findings, showing that HCV 1b infections breached a fast exponential growth from 1991 to 1998, while the HCV 2a infections had a fast exponential growth that occurred in around 1996–2001. Overall, this investigation helps to better understand HCV transmission in China and supports improvements of HCV prevalence control.
H
The earliest localized HIV-1 epidemic in China was widely accepted to occur in an IDU population from bordering provinces such as Yunnan and Guangxi in the late 1980s, 3 and then, the HIV-1 strain (Thai-B) was introduced into inner land through heroin trafficking. 4 In the early to middle 1990s, unregulated commercial plasma collection, which was common in the central Chinese provinces, such as Henan, Anhui, Hubei, and Shanxi, caused the outbreak of HIV-1 infection in the former blood donors (FBDs) in these regions. Due to shared transmission route with HIV, the coinfection of HCV and HIV is frequently observed in FBD patients in central China. 5
It was established that Southeastern countries (mainly Thailand) were the transmission source for HIV-1 in Chinese FBDs and Thai-B is the predominant HIV-1 strain in this population. 4 Although the introduction and outbreak of HCV infection in FBD may occur together with HIV, the transmission source of HCV in this population still remains unclear. HCV displayed a high genetic diversity and at least six major genotypes and more than 70 subtypes were identified worldwide. 6 The prevalence of HCV varies depending on the country and region. 7 –9 The most commonly detected genotypes in China are 1b and 2a, which accounted for more than 90% HCV infections. 10 The conducted study in Henan-derived FBDs indicated that 1b and 2a are also the dominant genotypes. 11 However, 3a, 1b, and 6a are the most common genotypes of HCV in Thailand and 2a was rarely found in Southeastern countries around China. 12 Therefore, it is highly unlikely to link the origin of 2a in Chinese FBDs to Southeastern countries, which suggested versatile origins of HCV in FBDs in contrast to HIV. Therefore, FBDs with HIV-1/HCV coinfections may represent a unique population, which can be used to study the coevolutionary history of HIV-1 and HCV in human.
In the present study, we recruited HIV/HCV coinfected patients from Fuyang city of Anhui province to perform phylogenetic analysis of HCV-NS5B and HIV-pol gene sequences. To investigate the transmission resource of HCV infection and characterize its evolution pattern in Chinese FBDs, we retrieved all available China-derived HCV NS5B sequences from the HCV database and performed phylogeographical analysis together with the Fuyang-derived NS5B gene sequences. Our findings provided new insights into the understanding of the transmission and evolution of HIV and HCV coinfections in China.
One hundred one FBDs infected with HIV-1, confirmed by Western blot analysis at the local Center for Disease Control and Prevention (CDC), were enrolled in March 2009 in Fuyang city. All participants had no obvious direct epidemiological linkage, such as familial or cluster infection relationships. Forty-seven of 101 HIV-1 patients were excluded due to ongoing antiviral therapy and the remaining 64 were included for the study. The mean age of 64 participants was 41.4 years and males accounted for 44.3% of the patients. Details on general patient information and blood donation history were collected by trained and qualified staff using a standardized questionnaire. The study was reviewed and approved by the Ethics Review Committee at the Minhang Hospital, Fudan University. Written informed consent was provided by all participants.
Whole blood specimens were collected in sterile tubes using EDTA as the anticoagulant. All samples were transferred to a laboratory within 12 h after collection. Separated plasma samples were stored at −80°C for viral RNA extraction. All plasma specimens were tested for the presence of HCV using the enzyme-linked immunosorbent assay (ELISA) kit (Kehua Biotech Co., Shanghai, China), and positive samples were confirmed by a third-generation ELISA kit (Abbott Diagnostics, Santa Clara, CA). Forty-six of 64 HIV patients were identified to be positive for anti-HCV antibodies and considered as HIV/HCV coinfections.
Viral RNA was extracted from plasma of HIV/HCV coinfections using the QIAamp Viral RNA Mini Kit (Qiagen, Inc., Valencia, CA) followed by cDNA synthesis using random primers with SuperScript III First-Strand Synthesis System (Invitrogen Corp., Carlsbad, CA). A 1,171-bp fragment in the HIV-1 pol gene, including the complete protease gene and the first 230 amino acids of the reverse transcriptase gene, was amplified by nested PCR amplification as previously described. 13,14 A 388-bp fragment in the NS5b gene of the HCV genome was amplified by nested PCR as previously described. 13 All PCRs were carried out with negative controls in a designated PCR clean room. PCR products were subject to sequence analysis by cycle sequencing and dye terminator methods with DNA Sequence Analyzer ABI 3730xl (Applied Biosystems, Inc., Foster City, CA). A total of 23 samples were achieved in obtaining both pol and NS5b gene sequences. Their sequences are in the process of deposition into the GenBank.
Individual sequence fragments for each PCR product were assembled and edited using the Sequencher 4.2 (Gene Codes Corp., Ann Arbor, MI). After managing adjustment, NS5b and pol sequences were aligned with reference sequences recommended by the HCV/HIV database using Bio-Edit version 7.6, respectively. Phylogenetic trees of NS5b were constructed by the neighbor-joining method using Mega 6.01, and the reliability of branching orders was assessed by bootstrap analysis using 1,000 replicates. Among 23 samples from patients with HCV/HIV coinfections, phylogenetic analysis of NS5b showed that 12 (52%) were subtype 1b, while 11 (48%) were subtype 2a (Fig. 1). Phylogenetic analysis of pol gene sequences showed that they were all HIV-1 subtype B (data not shown).
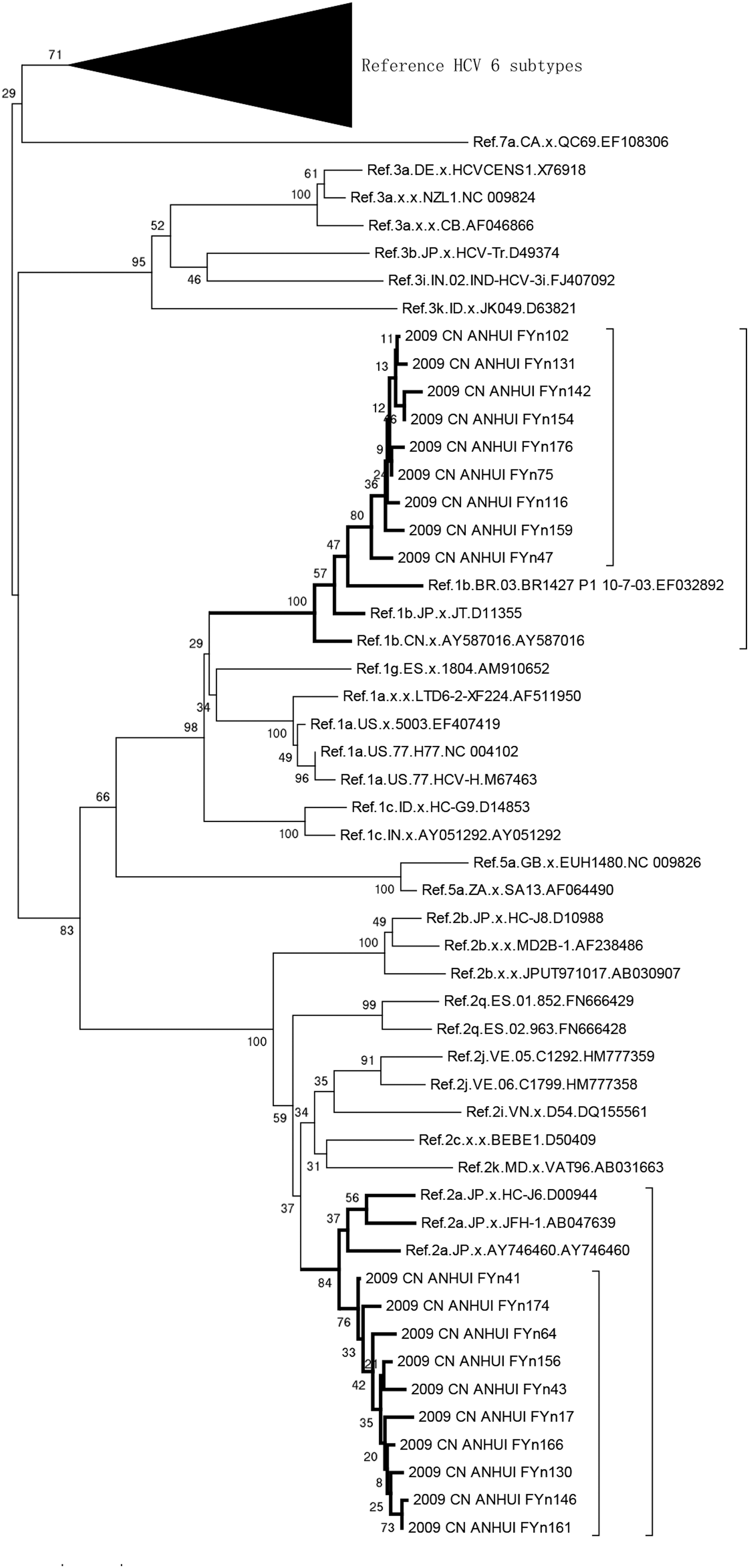
Phylogenetic tree analysis of 19 HCV sequences. A neighbor-joining tree of 19 HCV sequences and HCV reference sequences (retrieved from the HCV database) was constructed by using Mega 6.02; nine sequences formed a cluster with Ref. 1b subtype with 100% bootstrap probability; 10 sequences were clustered with Ref. 2a subtype with 84% bootstrap probability. HCV, hepatitis C virus.
To trace the transmission resource of HIV and HCV into FBDs in China, data sets of Anhui sequences (HCV1b NS5b, HCV 2a NS5b, and HIV pol) (Table 1) and retrieved Chinese sequences from public HCV database (Supplementary Table S1; Supplementary Data are available online at
HCV, hepatitis C virus.
The MCC tree of HCV 1b subtype (Fig. 2a) indicated that two Chinese HCV clusters were formed at almost the same time (tMRCA: 1979) with one being the dominant strain responsible for the outbreak of HCV in FBDs, including HCV 1b isolates from Anhui, Henan, Shenyang, Yunnan, Beijing, and Shanghai. The other may represent spatial infections, including the strains only from Hubei. Notably, the small cluster of HCV 1b displayed the close transmission linkage between Chinese strains and Japanese strains. In the MCC tree of HCV 2a (Fig. 2b), unlike the HCV 2a in FBDs, the whole Chinese HCV strain cluster was supported with low probability (0.15), which suggested the heterogeneous population of Chinese HCV strains with various transmission routes. The divergence tMRCA of HCV 2a in China was estimated in 1990, 11 years (SD 2.9 years) later than the most recent date for HCV 1b samples collected. In the MCC tree of HIV pol (Fig. 3), one major cluster formed in around 1988 corresponding to the outbreak of HIV in FBDs. In general, the transmission resource of HIV-1 in Chinese FBDs is undoubtedly Thailand, while the transmission resource for HCV is still not so confirmative. Japan rather than Thailand is more likely to be the origin of Chinese HCV in FBDs, however, the linkage between China and Japan is also very weak. These results not only suggested that HCV and HIV were introduced into FBDs at different times from different resources but also indicated the distinct transmission patterns between the two HCV subtypes.
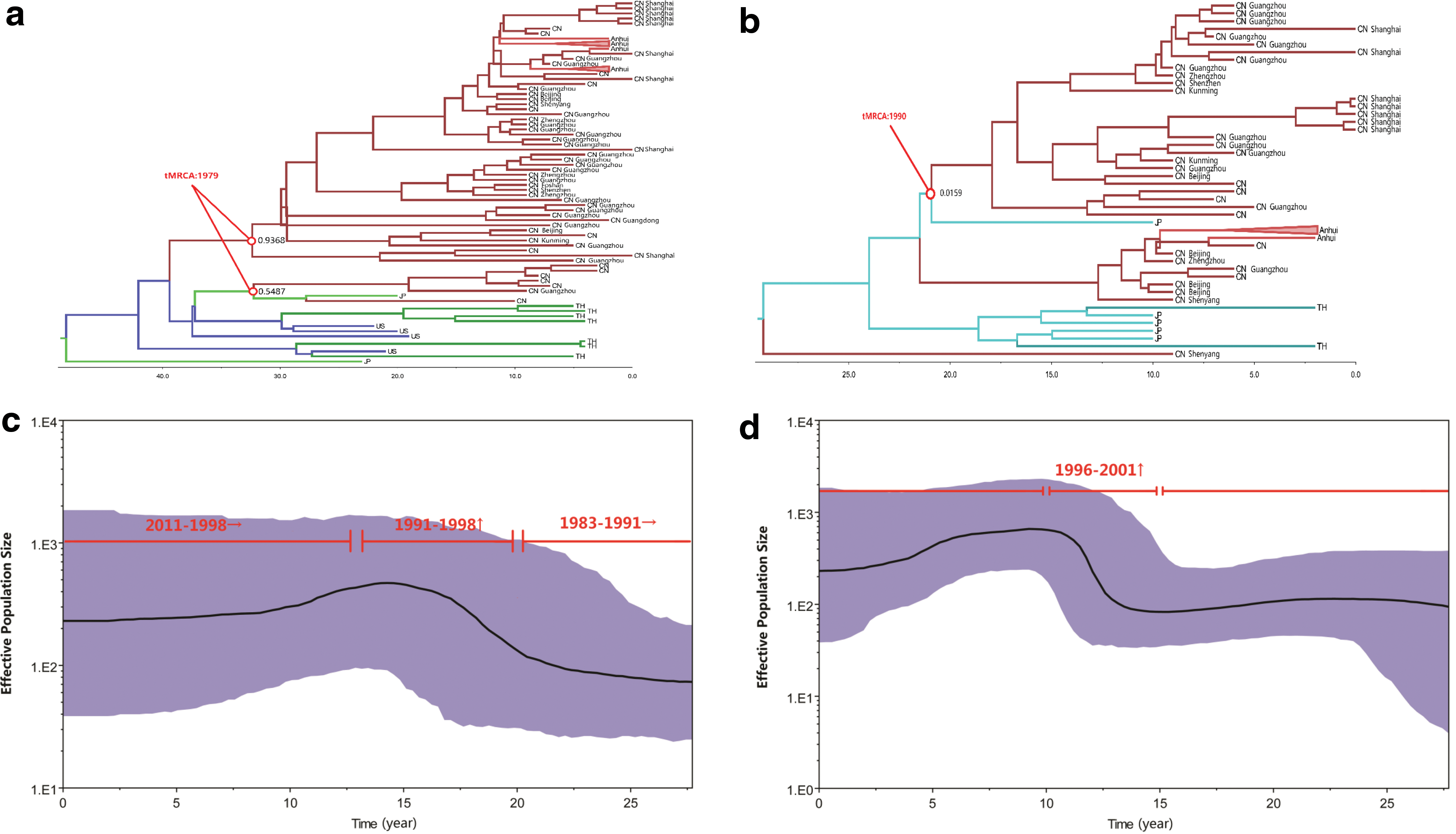

MCC tree analysis of HIV-1. In addition to the 19 HIV-1 isolates presented in Table 1, sequences received from HIV databases were also included. Branches were colored according to their respective geographic origins. Posterior probabilities were shown at the respective nodes, while the estimated time points of the origin for the nodes were noted by red circle. Below the tree is a time scale from 1969 to 2011, which measures each subtype origin and evolution. Color images available online at
To show the epidemic history of HCV, we used Bayesian to reconstruct the Bayesian Skyline Plot (BSP). Through the MCMC process, the BSP generalizes skyline plots from the targeted sequence data set, samples the distribution of the generalized skyline plots, and combines the plots to yield a posterior distribution of effective population size through time (Fig. 2c, d). Since credibility intervals are provided for effective population size at every point in time, from the present back to the most recent common ancestor of the sampled sequences, a history of HCV-infected population size is inferred. 19 Figure 2c and d shows the estimated BSPs of HCV 1b and 2a genotypes based on relax-exponential models, respectively. These are flexible, nonparametric estimates of past changes in population size, 19 plotted using the “log” files and “trees” files resulting from the coalescent procedures. For the HCV 1b, a relatively constant population size was maintained from 1983 to around 1991, and then, a fast exponential growth was breached from 1991 to 1998 (Fig. 2c). Subsequently, the growth gradually slowed and has remained slightly upward till present. For the HCV 2a, fast exponential growth occurred in around 1996–2001, which was later than that of HCV 1b and displayed a smaller scale (Fig. 2d). Particularly, the rapid growth from 1991 to 1998 overlaps a period when contaminated plasma collection was common in China, 20 and the abrupt slowing around 1998, however, was concurrent to a time point when the Chinese government outlawed the use of paid blood donors, which went into effect that year. 10,21 –23
Fuyang city lies in the east of Anhui province and is neighboring to Henan province, which is the most severe FBD infection province in China. More than half of FBD infections of Anhui province are from Fuyang city and there are high frequencies of HIV-1/HCV coinfection in FBD HIV-1 patients of Fuyang. 11 The prevalence of HCV infection (72%) among HIV-1-positive patients was similar to the coinfection rate in FBDs reported previously in Hubei (78.6%), Shanxi (85.0%), and Henan provinces (82.2%). 10,20,24,25 Phylogenetic analysis showed that all patients were infected with HIV-1 subtype B and that only HCV subtypes 1b and 2a were detected in coinfection cases. FBDs are the main susceptible population for the transmission of HCV subtype 1b and the latter formed a big cluster and showed very close linkages, consistent with the explosive transmission of HCV 1b during the process of unregulated commercial blood donation. 20,26 HCV 2a was introduced into FBDs in a later phase than HCV 1b, and therefore, the former did not form any significant cluster in these populations. Notably, various HCV genotypes may lead to different disease progression, and HCV 2b was believed to be associated with more severe syndrome and prognosis. 27 Consistent with this notion, our results showed that HCV 2b displayed faster evolutionary rates than that of HCV 1a in FDB with coinfections. Meanwhile, the more rapid evolutionary rate facilitated an expanding transmission of HCV 2a into various populations except FDBs, including illegal drug users and sexual transmission, which eventually led to the fast growth of HCV 2a infection in China.
Since there are limited data to investigate the transmission resource of HCV into Chinese FBDs, we focus our study on the origin of HCV 1b and HCV 2a strains in contrast to HIV. It was established that HIV infections in FBDs are introduced from Thailand, however, two HCV genotypes strains showed distinct origin time and transmission resource in contrast to HIV. By the phylogeographical analysis, Japan rather than Thailand is more likely to be the transmission resource for HCV. Meanwhile, two HCV genotypes in one population (FBDs) suggested multiple introductions into this population. Indeed, multiple clusters formed by Chinese strains also confirmed this possibility. Due to the absence of early HCV strains, we cannot exactly estimate the origin of Chinese HCV 1b or HCV 2a strains, which may hinder the understanding of HCV transmission pattern in China. Our results first revealed the versatile transmission resource of HIV and HCV, even various HCV genotypes.
In summary, this study identified the origin and transmission pattern of two HCV genotypes and HIV in patients with coinfections. The knowledge is critical to control the prevalence of HCV and highlights the need to increase education regarding sterile techniques and equipment for blood donation as well as to develop methods to improve treatment outcomes.
Footnotes
Acknowledgments
We are indebted to the patients for their participation and the staff at the local CDC for data collection and laboratory testing. This work was supported by the National Natural Science Foundation of China (81273136 and 81572395).
Authors' Contributions
Z.M. conceived the study, carried out the molecular genetic studies, and drafted the manuscript. L.D., J.W., and P.Q. participated in the design of the study and performed sequences analysis. R.X. and Y.N. coordinated the study, participated in the experimental design, and helped to draft the manuscript. J.X. proposed the concept of the study, designed the study, formulated the major conclusion, and revised this manuscript. R.H. provided critical discussion and revised the manuscript. All authors read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.