
Abstract
Antiretroviral therapy (ART) can be compromised by selection of drug resistance strains, which can be promoted by lack of adherence during therapy and drug tolerance, and some of these drug-resistant strains can persist for years as minority populations. The K103N drug resistance mutation is selected by the use of non-nucleotide reverse transcriptase inhibitors, including nevirapine or efavirenz (EFV), used in low-income countries. Here we describe the use of a less expensive qualitative point mutation polymerase chain reaction (PMqPCRK103N) targeting K103N mutation. To validate the use of this methodology, we tested previously sequenced samples from patients treated with highly active ART with viral loads above 2,000 copies/ml and compared the results of our assay with Illumina deep sequencing. Due to its low cost and high specificity, this test is particularly suitable for low-income countries to screen for pretreatment resistance in patients either initiating ART or failing first-line regimens containing EFV.
Introduction
A
The WHO recommends a first-line regimen based on two nucleotide reverse transcriptase inhibitors (NRTI) and a non-nucleotide reverse transcriptase inhibitor (NNRTI) in resource-limited settings. When patients taking this first line start to fail therapy, the first mutations to appear are NNRTI related, such as K103N and Y181C. K103N deserves special attention since it does not substantially affect viral fitness, can be transmitted with high efficiency, and is retained in the viral genome for more than 3 years postinfection. 3,7,14
In addition, K103N is one of the most common drug resistance mutations (DRMs) in most low-income countries as nevirapine (NVP) or efavirenz (EFV) are the first drugs adopted. 15 NVP has been extensively utilized as monotherapy during labor to prevent mother-to-child transmission, 13,16 accompanied by a week of zidovudine (AZT) and lamivudine (3TC) treatment postpartum. Nonbreastfeeding newborns usually receive (NVP) or (AZT) for 6 weeks after birth. 15 These treatments select for low-frequency HIV drug resistance variants that are not easily detected by Sanger-based methods. 13
Despite the relative low sensitivity and high cost of Sanger-based sequencing, it still remains the dominant platform for drug resistance studies. This method has good specificity and can accurately detect critical DRM codons. However, it is unable to detect the low-frequency variants that are present in <5%–30% of the virus population in the sample, 13,17 –23 and its sensitivity to identify DRM is <70%. 24 –26 Sanger sequencing is also unable to discover complex combinations of mutations 24,27 present in quasispecies from a single sample. These limitations can compromise genotyping, affecting treatment decisions. 20,28 In 2014, both Bellecave et al. 20 and Lipscomb et al. 28 presented patients with low-frequency DRM previously detected to antiretroviral (ARV) treatment, compromised by the emergence of those viruses.
Quantitative polymerase chain reaction (qPCR) is an alternative method to detect low-frequency mutations in virus populations.
13,16
qPCR genotyping is potentially more sensitive, specific, and easy to perform than Sanger sequencing. In fact, qPCR is especially useful for screening large numbers of patient samples, having been incorporated since the early 1990s for the quantification of HIV-1, a predictor of AIDS progression.
17,29
Currently, qPCR is considered the gold standard for virus detection and quantification,
30
also accurate and reliable for detection of point mutations in human genome as well as in infectious agents.
31
–33
Although qPCR methodology has the sensitivity and specificity to detect point mutations in HIV-1, its performance can be inhibited by sequence variation in the virus present in primer- and probe-binding regions. This is critical for genotyping HIV-1 group M viruses since the nine major subtypes and at least 71 Circulating Recombinant Forms (
Herein, we targeted K103N, one of the main DRMs selected during NNRTI treatment, 13,15,16 and developed a point mutation qPCR method targeting K103N (PMqPCRK103N) with SYBR Green. This method can be adopted in low-income countries without losing sensitivity and specificity when compared to Sanger-based methods. 13,16 We have defined PMqPCRK103N specificity and sensitivity in a well-characterized HIV-1 isolate panel composed of drug-experienced individuals infected with subtypes B, C, and F by comparing this assay to sequencing data generated by Illumina deep sequencing, a methodology able to detect mutations as low as 2%. 37 –41
Materials and Methods
We used 103 plasma samples (87 collected from 2003 to 2006 and 16 in 2015) from Brazilian patients infected with HIV-1 subtypes B, BF, F, and C, on antiretroviral treatment. All samples were selected from patients with previous Sanger sequencing analysis and had viral loads greater than 2,000 copies/ml, indicating treatment failure or loss of immune control. Twenty-eight of the seventy-five patients failing highly active antiretroviral therapy (HAART) had no K103N mutation identified by Sanger. We designed a sensitive qPCR test to detect a K103N mutation associated with NNRTI, for which we adapted primers already described
13,16
to better match HIV-1 sequences circulating in Brazil (see Supplementary Table S1 for details; Supplementary Data are available online at
Preparation of virus template
HIV viral RNA was extracted from plasma with the QIAamp® Viral RNA kit (QIAGEN®). The cDNA template for our experiment was obtained with Superscript III (Life Technologies) with random primers according to the manufacturer's instructions. We then performed an external PCR, to ensure enough DNA for repeat testing, with primers (P1 and P2), previously described by Johnson et al., 13 which amplifies from nucleotide 1,799 to nucleotide 2,850 relative to the HXB2 sequence (Supplementary Table S1). Approximately 30 ng of the cDNA was added to the following PCR mastermix in a first round of PCR amplification: Taq Gold buffer 1 × , MgCl2 0.5 mM, dNTP 250 nM, Primers P1 Fw, P2 Rev 250 nM each, and Taq Gold Invitrogen 1 U. The cycling conditions were as follows: 1 cycle of 94°C 5 min; 6 cycles of 94°C 1 min, 55°C 1 min, 72°C 1.20 min followed by 35 cycles of 94°C 1.20 min, 55°C 1 min, 72°C 1.20 min, and 1 cycle of 72°C 7 min. The approximate 1,000 bp amplicon was observed by gel electrophoresis in a 1% agarose gel stained with ethidium bromide. The amplified product was purified with Montage PCR centrifugal filter devices (Millipore Corp.) and quantified on a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.) before joining the second round on the PMqPCRK103N.
PMqPCRK103N
The objective of the test requires the amplification of the same region with two different sets of primers. The first set of primers (general target amplification for mutant and nonmutant detection) anneals in a constant region of the RT gene, exhibiting low diversity between resistant and wild-type viruses. General target amplification is necessary to obtain the baseline Ct value corresponding to the Ct value that represents all templates regardless of mutation status. For the general amplification, we used one forward primer (K103N Fw) and two reverse primers (K103N Rev Tot1 and K103N Rev Tot2) to amplify all HIV-1 variants circulating in Brazil. The cycle threshold detected with these general primers generates Ct1. For the questioned K103N DRM (specific target amplification), we used the same forward (K103N Fw) and four reverse primers (K103N Rev AAT1, K103N Rev AAT2, K103N Rev AAC1, and K103N Rev AAC2) similar to the first set but with one located nucleotide downstream (5′→3′) of the general reverse primer (Fig. 1). With this second set of primers, we obtained the Ct2 representing the presence of the K103N mutation. It is necessary to calculate the ΔCt value (Ct2 − Ct1) to determine the presence or absence of the K103N mutation. All samples were tested in triplicate reactions with the Ct average used to calculate the final ΔCt. We expected to have low ΔCt values for the samples with the questioned DRM and higher values for those without.

Scheme representing primers annealing with coordinates relative to HXB2. P1 Fw and P2 Rev—forward primer and reverse primer, respectively, K103N Fw—forward primer used in qPCR, K103N Rev Tot1, K103N Rev Tot2—reverse primers used with K103N Fw for total detection; K103N AAC1, K103N AAC2, K103N AAT1, K103N AAT2 second set of primers used with K103N Fw for allele-specific mutation. qPCR, quantitative polymerase chain reaction.
Individual reactions were carried out in a total volume of 20 μl/well in 96-well PCR plates on an Exicycler TM Real Time PCR system (Bioneer Corporation) with SYBR Green reaction mix (Applied Biosystems). The reaction conditions were as follows: 10.0 μl of SYBR mix (Applied Biosystems), primer K103NFw 300 nM pM, primers RevTot1 and RevTot2 250 nM for all DNA templates or primers RevAAC1, RevAAC2, RevAAT1, RevAAT2 25 pM for K103N DRM, and 0.1 ng of target DNA per reaction. The cycling conditions were as follows: 95°C for 11 min followed by 35 cycles of 95°C 30 s, 57°C 1 min. and the melting curve was recorded from 55°C to 94°C increasing 1°C per minute to check the identity of amplified material.
PMqPCRK103N validation
To establish the sensitivity and specificity of the PMqPCRK103N, we used two clonal samples, one representing the wild-type virus (NL4-3) and the other carrying the K103N (clone 154) mutation. Ten-fold dilutions were produced starting with 1 ng/μl, ∼8.63 × 108 to 8.63 × 104 copies, of purified amplicons generated in the first amplification (P1 and P2 primers) for each clone to determine the best annealing temperature, primer concentrations, proper amount of sample for each assay, and to also establish the standard curve (Fig. 2).
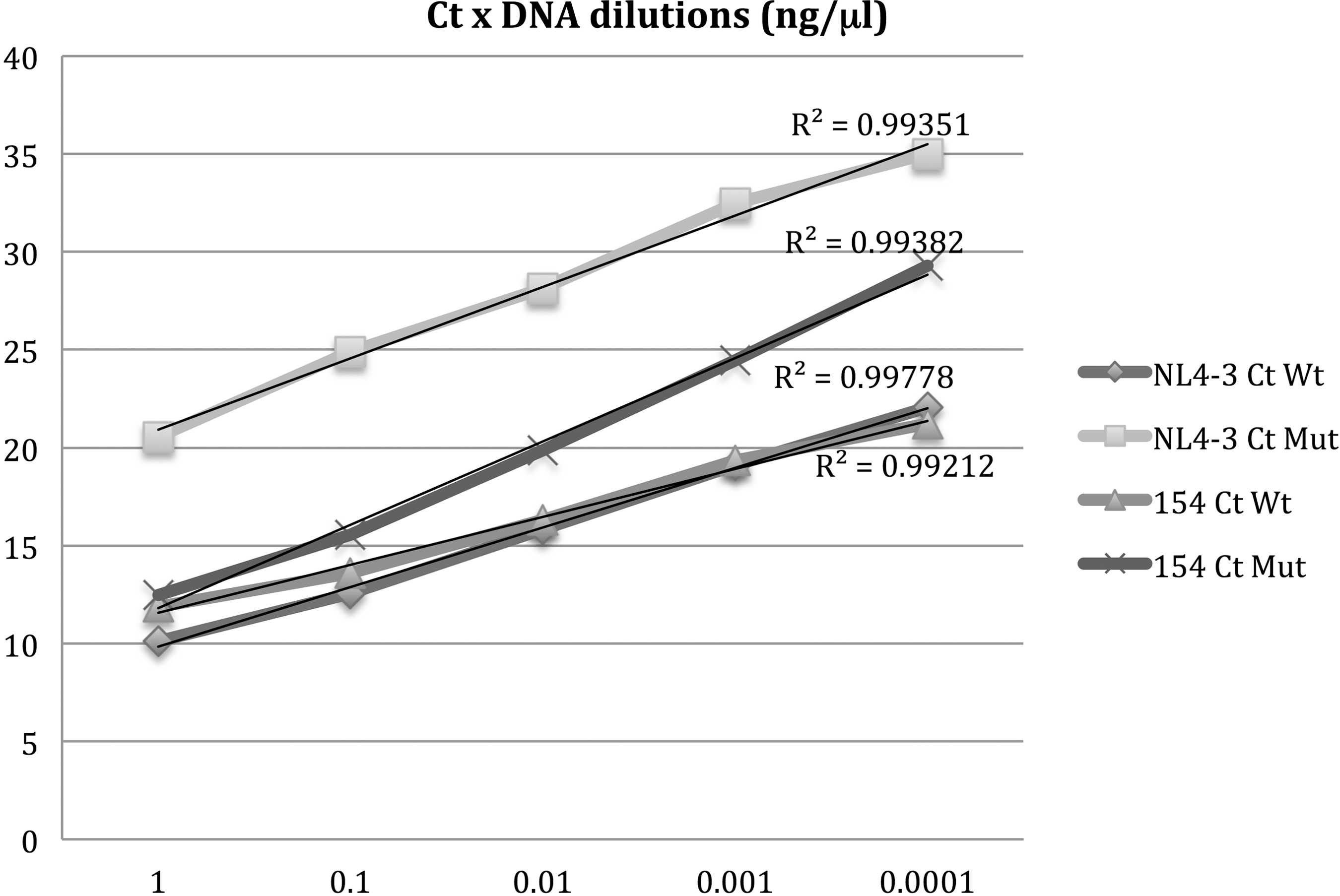
Ct × DNA dilutions. Results obtained NL4-3 and 154 clone samples in different concentrations. Average of total Ct is the value obtained with primers K103N Fw and K103N Rev Tot1 and K103N Rev Tot2; K103N Ct was obtained with K103N Fw and K103N Rev AAT1, K103N Rev AAT2, K103N Rev AAC1, and K103N Rev AAC2 in triplicate experiments.
Illumina MiSeq sequencing
The 1,067 bp PCR product generated with the P1 and P2 primers was subjected to Illumina deep sequencing. The DNA was either purified with AMPure XP beads (Beckman Coulter, Inc.) or gel purified with a MinElute Gel Extraction Kit when there were PCR products in addition to the expected 1,067 bp PCR product (Qiagen). Paired-end DNA libraries were prepared with the Nextera XT DNA sample prep kit (Illumina) according to the manufacturer's protocol. Briefly, ∼1.0 ng of PCR product was subjected to tagmentation with Nextera amplicon tagment enzyme mix at 55°C for 5 min followed by a twelve-cycle PCR with the Nextera PCR mix to add Illumina indexes to the PCR products. The reaction conditions were 72°C for 3 min, 95°C for 30 s followed by 12 cycles of 95°C 10 s, 55°C 30 s, 72°C 30 s and finally one cycle of 72°C for 5 min. The average size of the fragmented libraries was 683 bp, later purified with AMPure XP beads (Beckman Coulter, Inc.) at a 5:3 ratio of PCR product to bead. The product of each sample was normalized to 2 nM and pooled together in preparation for loading into the Illumina MiSeq Sequencing cartridge for sequencing. Samples were sequenced with an Illumina MiSeq Sequencing 500-cycle version 2 kit (Illumina).
Illumina MiSeq data analysis
The next-generation sequencing (NGS) data generated with Illumina for each sample were analyzed with Geneious Pro Version 7.1 (Biomatters). From a total of 103 sequenced samples, 21 had low coverage (<100 reads) and were excluded from the study. Next, we randomly subsampled 15,400 sequences from each sample with Seqtk tool from the Geneious software. This downsampling reduced computational load while still providing 3,589 reads of average coverage of each base in the 1,060 bp amplicons. Reads were then mapped to the HIV-1 reference sequence HXB2 (GenBank NC_001802) annotated with DRMs in accordance with the “medium” preset sensitivity mapping parameters in Geneious. Polymorphisms (defined as variants found in at least 2% of reads, supported by at least five reads, and with a p-value of at least 10e-6) at the K103N resistance site were identified with the Geneious genotyping tool. A workflow automating these steps is available on request. Samples with the K103N mutation found below 2% were considered negative.
Statistical analysis
The data from Geneious identified which samples contained the K103N mutation and at what frequency were exported to Microsoft Excel 2013, and the plots were generated with R v3.02 in RStudio v0.98. We established sensitivity and specificity of PMqPCRK103N with the Clinical Research Calculator on VassarStats statistical computation website (
Results
Initially, we designed standard curves with five different concentrations ranging from 1.0 to 0.0001 ng/μl of NL4-3 or 154 cloned strain samples, and the efficiency of the assay was 99.768% (slope of −3.328 and r
2 = 0.993
We initially separated the samples into two groups: those with K103N DRM and those with mutations other than K103N (NR-DRM), based on the genotyping generated by Sanger sequencing (Tables 1 and 2). As mentioned before, the Sanger sequencing method has a detection limit of minority populations around 5%–30%,
13,17
–23
and so, we decided to enlist Illumina MiSeq to investigate the limitations of our PMqPCRK103N. From the initial 103 samples, we obtained good Illumina sequence data in 82 samples and only these sequences were adopted for further analysis. We used data generated with Illumina MiSeq for each sample as our gold standard to determine the presence or absence of the K103N mutation and the total of mix populations to evaluate our PMqPCRK103N performance. All 47 samples carrying the K103N mutation based on both methodologies displayed an average of 82% of reads carrying K103N by Illumina sequencing (Range 5%–99.8%). The cutoff calculated obtaining ΔCt average of all positive samples was 2.5. To calculate the cutoff value for ΔCt, we added two standard deviations (2.12) resulting in a cutoff of 6.742 Cts (see Fig. 3 for details). There was a good correlation between the percentage of reads carrying K103N by Illumina sequencing and the observed ΔCt. However, we witnessed outliers with low and high amounts of reads and ΔCt. When the 11 specimens originated from patients failing ARV regimens containing previous NNRTI and no K103N mutation by Sanger methodology, we realized that two of them, 044/05 and 012/06, contained the K103N mutation frequency at 4.3% and 31.3% by Illumina MiSeq, respectively. We considered one sample, 059/05, as undetermined, although the ΔCt value was below the cutoff value. The reason for this is because the K103N frequency achieved with Illumina MiSeq was under 2.0%, this value was considered low for reliability. When all NR-DRM samples were analyzed, we found all of them had ΔCt values below the cutoff value, thus suggesting the presence of the K103N mutation. Remarkably, all these samples were from subtype C isolates circulating in Brazil, and the percentage of reading carrying K103N was below the cutoff value of Sanger methodology except for samples 457 and 462, which had 33.3% and 96.2% of readings with K103N DRM, respectively. Considering Illumina sequencing as the gold standard, the specificity of the PMqPCRK103N assay was 94.53% and 91.98% for sensitivity (

Mean and distribution of ΔCt (ordinate) results obtained for samples with K103N mutation and NRDRM. N is for number of samples; NDRM, no related drug mutation.
qPCR, quantitative polymerase chain reaction.
Bold values indicate indeterminate and negative results for K103N mutation.
IND is for inconclusive result if compared to Illumina MiSeq. NR-DRMs are samples from patients failing HAART carrying mutations other than K103N.
DRM, drug resistance mutation; HAART, highly active antiretroviral therapy.
To analyze the reasons for the discrepancy between the amount of reads containing K103N and the ΔCt values, we aligned the sequences with different ΔCt values exhibiting the region of forward and reverse primer anchorage (Table 3). Of note, there were much more mismatches, in 3′ primer positions, between the forward and reverse primers in samples with ΔCt value discrepancies when compared to the concordant counterpart.
Based on our cutoff, samples with ΔCt <8.814 have K103N mutation present. “Bold” nucleotides show the discrepancies against the primer. Sample 093/05 was not detected by Sanger methodology.
Discussion
Herein, we designed a low-cost, highly sensitive qPCR test based on previous works 13,16 to detect K103N, an important point mutation associated with (EFV) and (NVP) resistance. The estimated cost of our assay here in Brazil is US$2.00 to US$5.00 per sample, about US$10.00 less than Sanger sequencing. The cost difference, in part, is attributed to embedded taxes. In addition, the time spent in real-time execution is lower by at least 8 h. Due to the sensitivity limitations of Sanger-based sequencing, we decided to use Illumina sequencing to better investigate the amount of mixed populations to check the performance of our PMqPCRK103N. In brief, we evaluated HIV RT sequences from 66 Brazilians harboring subtypes B, BF, F, and C. We designed PCR primers accommodating all polymorphic sites located at codon position 103 with a Brazilian sequence database. Utilizing this strategy, we were able to generate suitable primer sets to detect K103N mutations in the background of Brazilian HIV-1 isolates. In our hands, this methodology was better performed with amplicon concentrations above 0.01 ng/μl (around 107 copies). We tried to evaluate our cutoff value for ΔCt considering 47 selected samples, where Illumina sequencing yielded results presenting K103N proportions above 2%. Although Illumina NGS technology has the lowest error rate compared to the other available NGS platforms, we prefer to use conservative profiles for demand polymorphisms. In this case, we used 2% based on the assay sensitivity of the NGS technology. Afterward, an assay cutoff was generated and utilized to check unknown samples from other individuals.
Since we utilized the product of PCRs from first-round PCR with external primers as DNA input on our PMqPCRK103N, this amount of DNA is easily obtained starting with viral RNA extracted from plasma from HIV-1 patients above 2,000 copies/ml, following the Brazilian Ministry of Health protocol. 42 Unfortunately, we do not have information regarding the drug regimen of patients, only that they have been treated according to the Brazilian Ministry of Heath 42 protocol with TDF, 3TC, and EFV (Supplementary Table S2).
Analyzing the 82 samples that had good coverage data through Illumina sequencing, we were able to identify isolates carrying K103N DRM applying an independent method. We also adopted Illumina data as our gold standard to calculate our sensitivity and specificity. Overall, the prevalence between PMqPCRK103N and Illumina MiSeq was 99.92% for the presence of K103 N DRM.
In our analysis, we treated ΔCt as a qualitative variable that is proportional to identify the presence of the K103N mutation. We calculated the ΔCt test cutoff as 6.742, allowing us to discriminate samples carrying K103N from the wild-type counterpart. We established that one sample (059/05) was undetermined for the questioned mutation as the K103N frequency was 1.5% in Illumina Deep Sequencing with a ΔCt lower than the cutoff of our test. Of note, this sample had only lysine in RT position 103 when sequenced by Sanger methodology.
We have selected some samples showing concordant and discordant ΔCt values with the proportion of K103N reads in our NGS methodology as well as designing an alignment with the HXB2 reference sequence and our forward and reverse primers (Table 3). We have observed a high number of mismatches in 3′ primer positions, between the forward and reverse primers in samples demonstrating a discrepancy in ΔCt values when compared to the concordant counterpart. Mismatches at primer 3′ ends are likely to modify the initial PCR causing lags in the amplification chain reaction yielding a higher Ct. This fact may explain the disparity in results where samples carrying a high proportion of K103N reads had high ΔCt values such as isolates 193/05, 058/06, 011/06, and 072/06 as opposed to samples 027/06, 104/06, 045/05, and 177/05.
We suggest that before adopting this methodology, the ΔCt cutoff value with the samples circulating in the country carrying the K103N mutation, as well as wild-type sequences from drug-naive individuals to better fit the primers with circulating viral variants, should be calculated. In our case, we think it may have superior results if we develop new forward primers with some more degenerate nucleotides to better accommodate polymorphism present in these sequences (Table 3).
We did not compare our allele specific polymerase chain reaction with other groups, but Jonhson et al. 13 reported sensitivity variation from 96% to 99% with specificity up to 99%. Pérez et al. 16 inform only that the lower limit of K103N DRM was 0.1%. Our test incorporated SYBR Green instead of TaqMan to minimize costs as well as primers, different from those developed by other groups, so as to match with HIV-1 circulating in Brazil (data not shown). Another point to be considered is that our assay was developed only to identify low-frequency mutations, but not to quantify as was described by other groups. 13,16
Some studies have reported that the importance of low-frequency DRMs and K103N is implicated to produce high-level resistance to EFV, 43 –46 both EFV and NVP known to select K103N DRM. 9,47 This DRM can also be found in children born from HIV-positive mothers under prevention of mother to child transmission interventions, as reported in a study in Kenya where it was the major DRM identified. 48 Due to its low viral fitness cost, 49 K103N can be present in DRM transmission cases. It is reported that virological failure may occur ∼6 months after being detected. 20 It has also been reported that patients with low-frequency DRMs ranging from 0.1% to 1.0% and with virus load above 2,000 copies/ml could lead to DRMs with stronger impact in treatment outcome. 8,19,44,50 This level of minor mutant detection is achievable by allele-specific real-time PCR technology. 50
Conclusions
We have developed an inexpensive qPCR incorporating SYBR Green to detect a K103N low-frequency mutation for the HIV-1 circulating in Brazil. We have used 103 samples of Brazilian patients infected with HIV-1 subtypes B, BF, F, and C and compared our results to those of both the Sanger-based method and MiSeq deep sequencing. Our results confirm that PMqPCRK103N can be useful as a qualitative test, especially as the first-line drug regimen, adopted in low-income countries, includes EFV or NVP plus Stavudine (D4T) or AZT. 51 Owing to the low negative predictive value of our PMqPCRK103N, we think that it is critical to associate new qPCRs targeting other key NRTI or NNRTI DRMs, such as M184V or Y181C, to increase the likelihood of identifying patients failing first-line regimens. As the test was developed as a proof of concept, other targets can always be developed taking into account the diversity of the circulating viruses in the region. Since the cost of the HIV genotyping test based on Sanger sequencing is very high, about 80% more expensive than qPCR and not accessible to low-income countries, we propose the use of our PMqPCRK103N for first-time screening in naive treatment HIV-1 individuals or in patients with virological failure in first-line antiretroviral regimens in low-income countries to minimize expensive HIV genotyping. This test will also reduce the early emergence of the DRM virus and increase the selection of patients for second-line regimens to suppress again patient viremia and diminish the costs of sequencing and identification.
Footnotes
Acknowledgments
This work was sponsored by CNPq, FAPERJ, CAPES, Department of AIDS/STD and Hepatitis, Brazilian Ministry of Health. English review and revision by Mitchell Raymond Lishan, native of Chicago, Illinois, USA-UCLA 1969.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.