
Abstract
Coreceptor switching from CCR5 to CXCR4 is common during chronic HIV-1 infection, but is even more common in individuals who have failed antiretroviral therapy (ART). Prior studies have suggested rapid mutation and/or recombination of HIV-1 envelope (env) genes during coreceptor switching. We compared the functional and genotypic changes in env of viruses from viremic subjects who had failed ART just before and after coreceptor switching and compared those to viruses from matched subjects without coreceptor switching. Analysis of multiple unique functional env clones from each subject revealed extensive diversity at both sample time points and rapid diversification of sequences during the 4-month interval in viruses from both 9 subjects with coreceptor switching and 15 control subjects. Only two subjects had envs with evidence of recombination. Three findings distinguished env clones from subjects with coreceptor switching from controls: (1) lower entry efficiency via CCR5; (2) longer V1/V2 regions; and (3), lower nadir CD4 T cell counts during prior years of infection. Most of these subjects harbored virus with lower replicative capacity associated with protease (PR) and/or reverse transcriptase inhibitor resistance mutations, and the extensive diversification tended to lead either to improved entry efficiency via CCR5 or the gain of entry function via CXCR4. These results suggest that R5X4 or X4 variants emerge from a diverse, low-fitness landscape shaped by chronic infection, multiple ART resistance mutations, the availability of target cells, and reduced entry efficiency via CCR5.
Introduction
H
Prior work from our laboratory has suggested that loss of entry fitness via CCR5 and increased CD4 binding may precede coreceptor switching. 3,41 Low nadir CD4 T cell count is a strong predictor of coreceptor switching, 37,42 as is a history of ineffective ART. 37,43 The increase in the frequency of coreceptor switching following ART could have trivial explanations such as a longer duration of infection, or it might be explained by changes in reverse transcriptase (RT) fidelity associated with drug resistance mutations 44,45 that increase env mutations, increased diversity of latently infected CD4 T cells from which X4 virus can be rescued, 46 or greater depletion of CCR5+ target cells. 32,47 –49 A long history of ART with only intermittent responses might lead to repeated seeding of the latent reservoir, 50 –52 providing more stochastic options for R5X4 virus archiving or generation.
These hypotheses prompted the current studies to examine both the evolution of env sequences and the entry competence via CCR5 or CXCR4 for full-length env clones isolated from nine treatment-experienced, viremic subjects selected from the SCOPE (Study of the Consequences of the Protease Inhibitor Era) cohort 37 just before and just after coreceptor switching. The results were compared to similar studies of 15 control subjects who maintained the same coreceptor use at the two sample time points. These studies are unique with respect to the short, 4-month interval between sample collection, the large number of unique full-length env clones with phenotypic entry data to correlate with sequences, and the extensive data on the status of the subjects enrolled in the SCOPE cohort.
Materials and Methods
Subject characteristics
Full-length env clones were amplified from plasma samples previously collected from 24 subjects enrolled in the SCOPE cohort with tropism results reported in studies by Hunt et al. 37 and Reeves et al. 53 Informed consent was obtained from all participants, and ethical approval was obtained from the ethics boards of each institution participating in the SCOPE at the University of California, San Francisco. Subjects were identified by code to allow linkage to clinical data, and the current study was approved by The Scripps Research Institute IRB (IRB 13-6137). Most subjects contributed two samples collected at roughly 4-month intervals; two subjects had four serial samples, and one subject had three serial samples. All subjects were classified into three groups according to their previous results from testing with an enhanced sensitivity tropism assay (21, 53) at the two (or more) sample time points as R5/R5, R5/DM (dual/mixed), or DM/DM. Table 1 presents data for each subject group. The R5/R5 (non-switch [NS] control) and R5/DM (switch [S]) groups were well matched for duration of HIV diagnosis, CD4 T cell counts, and viral loads. The two subjects in the DM/DM group had a longer duration of diagnosis, lower CD4 T cell counts, and higher viral loads than the other groups. All subjects were ART experienced and all had multiple drug-resistant mutations with poor or intermittent responses to therapy. None of the subjects had been treated with the CCR5 inhibitor Maraviroc. 18 All subjects were viremic at the time of sample collection.
Mean ± standard error CD4 T cells/mL.
Geometric mean (95% confidence interval) viral RNA copies/mL.
DM, dual/mixed.
Envelope cloning and coreceptor typing
Samples that had been polymerase chain reaction (PCR) amplified with primers containing a 5′ Mlu1 site and a 3′ Not1 site were received from Monogram. Samples were cut with Mlu1 and Not1 and ligated into pC1neo from Addgene. Colonies were picked from transformations of ligations and grown up and purified. Env clones were coexpressed with the NL4.3 env-negative, luciferase-positive reporter plasmid
54
in 293T cells (American Tissue Culture Collection CRL-3216). Coreceptor use of viruses or envelope clones was evaluated by infection of
Sequence analysis
Sequences were compiled, visualized, and aligned using Lasergene 8.1 software (DNASTAR, Madison, WI). To first rule out contamination or dual infection within each subject, we combined the env sequences obtained in this study with the HIV-1 2014 Web Alignment for env (3,551 sequences) from the Los Alamos National Laboratory HIV sequence database (
We also created amino acid alignments for each individual subject using MUSCLE within Geneious v8, followed by manual adjustment. Subject-specific maximum-likelihood phylogenetic trees were reconstructed from amino acid alignments using PhyML, 61 assuming the LG 62 amino acid replacement matrix, also within Geneious v8. Pairwise amino acid distance estimates were calculated for each person, including each set of sampling dates, using the LG matrix with the DIVEIN web server for sequence analysis. 63 For longitudinally sampled sequence sets, for each subject, amino acid divergence was estimated by comparing pairwise distance to the calendar time elapsed between sampling times. To assess the possibility of superinfection, sequences from subject 3,102 were aligned in Geneious v8 with 290 randomly chosen HIV-1 subtype B env sequences from the Los Alamos HIV database 2014 compendium. A neighbor-joining phylogenetic tree was generated. Sequence data will be deposited in GenBank on article acceptance.
Results
Subject characteristics
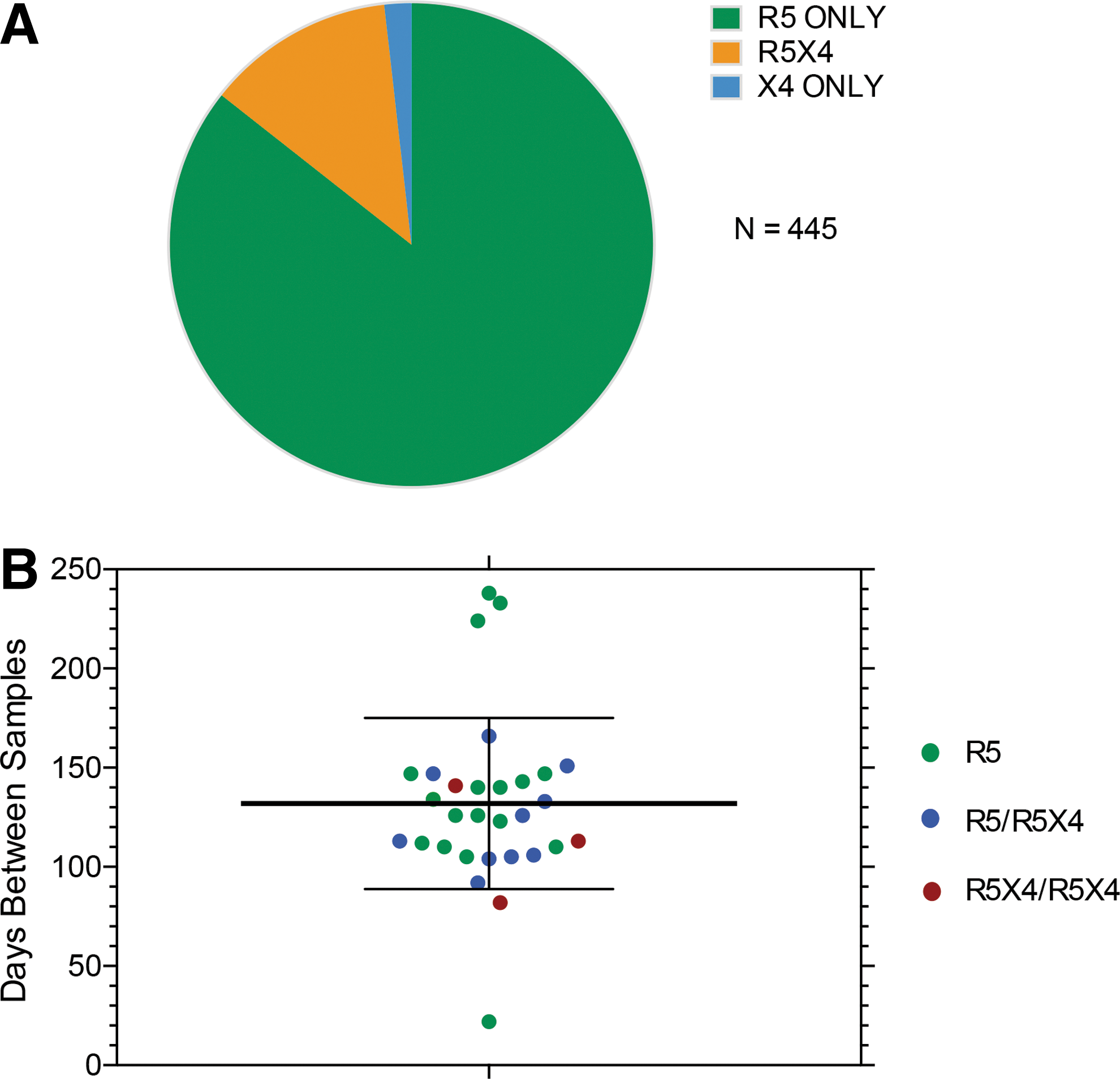
The interval between sample collection and the number of unique env clones with confirmed entry via CCR5 and/or CXCR4 are presented in Table 1. The interval between samples was slightly longer in the R5/R5 control group than the R5/DM or DM/DM groups (although this difference was not significant; Fig. 1), and the number of functional envs evaluated was lower in the R5/R5 group. This latter difference was a result of most env clones from the second sample in the R5/DM group mediating entry only via CCR5 (Fig. 1), so that evaluation of entry phenotypes continued until an R5X4 or X4 clone was identified. In only one subject where the second sample was originally typed as DM did we fail to identify an R5X4 or X4 clone after testing 151 functional and nonfunctional env clones. Since we had coreceptor entry data for unique env clones from each subject, the ambiguous DM classification was replaced by R5X4 or X4. Only 8/445 env clones from three subjects typed as X4, so the vast majority of unique clones from subjects with DM tropism results from populations of envs that were R5X4. We noted, as has been reported, 20 that a proportion of env clones from plasma were nonfunctional. All subsequent sequence analyses were performed only on the 445 env clones with confirmed entry function.
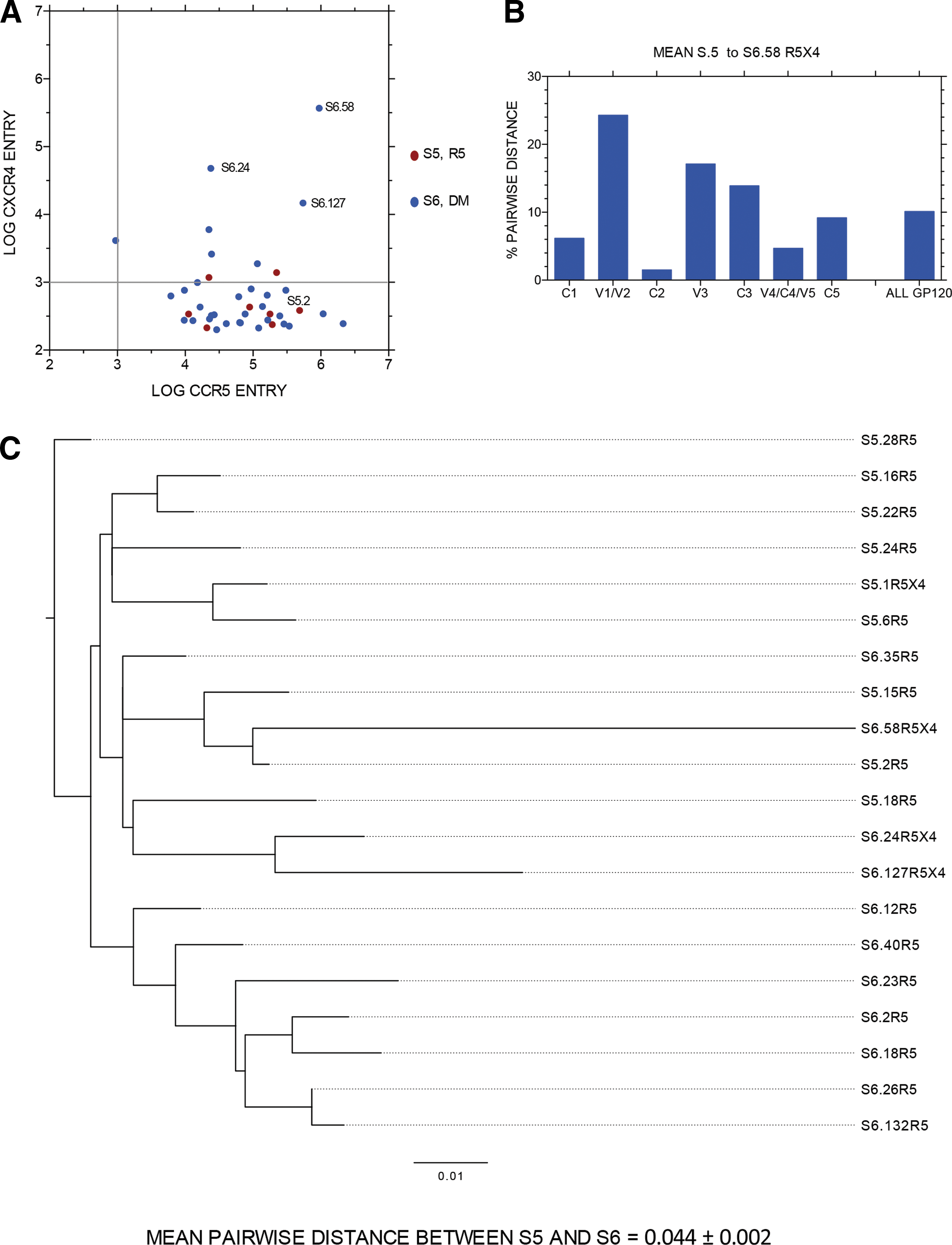
Evolution of env sequence and coreceptor use
Figure 2 presents data for one subject with R5 virus at the first sample time point (S5) and R5X4 virus 151 days later (S6). Figure 2A presents the entry data via either CCR5 or CXCR4 for each unique full-length env clone, with entry data presented as log10 RLU. Three env clones with robust R5X4 entry were identified in the second sample, and three more weak R5X4 clones and one weak X4 clone were also detected. Figure 2B shows the average pairwise distance in gp120 amino acid sequence from the first sample R5 sequences to R5X4 env clone S6.58, which conferred the most efficient entry via CCR5 and CXCR4. S6.58 was a genetic outlier (with no evidence of hypermutation). Hence, there was an overall, high (7.41%) divergence in all of gp120, with even more dramatic changes in V1/V2, V3, and C3. Figure 2C presents the phylogenetic tree of entire gp160 amino acid sequences from most functional env clones shown in Figure 2A. Note that S6.58 is most closely related to an R5 env clone from the prior sample time point, S5.2, whereas the other two robust R5X4 env clones (also outliers) were most closed related to each other and then to the R5 env clone S5.18.
Similar analyses were performed on functional env clones from all 24 subjects and are presented in the same format in Supplementary Figures S1–S24 (Supplementary Data are available online at
Figures 3 –5 present data for mean amino acid divergence (amino acid distance/time between samples) and mean pairwise amino acid distance between sample time points for full-length, functional env sequences for the three subject groups, now categorized as R5 to R5 (controls), R5 to R5X4 (switch), and R5X4 to R5X4 based on entry data for each env clone. One set of related sequences from one subject in the R5 to R5X4 group is omitted from the data because it represented superinfection or dual infection from a second source partner (Fig. 9, below). Figure 3 shows no significant difference in diversity or distance between the three groups, although the median value is slightly higher in the R5 to R5X4 group than the control group. Figure 4 shows the same data converted to diversity and distance per year to correct for differences in the interval between collecting sample 1 and 2. Again there is no significant difference between the three groups, although the two subjects (with three samples) in the R5X4 to R5X4 group show much higher values and the median values remain higher in the R5 to R5X4 group than the R5 to R5 controls. It should be noted that all env sequences from the two sample time points were included in the data presented in Figures 3 and 4, and the majority of env clones in the R5 to R5X4 group retained the R5 phenotype at the second time point (e.g., see Fig. 2).
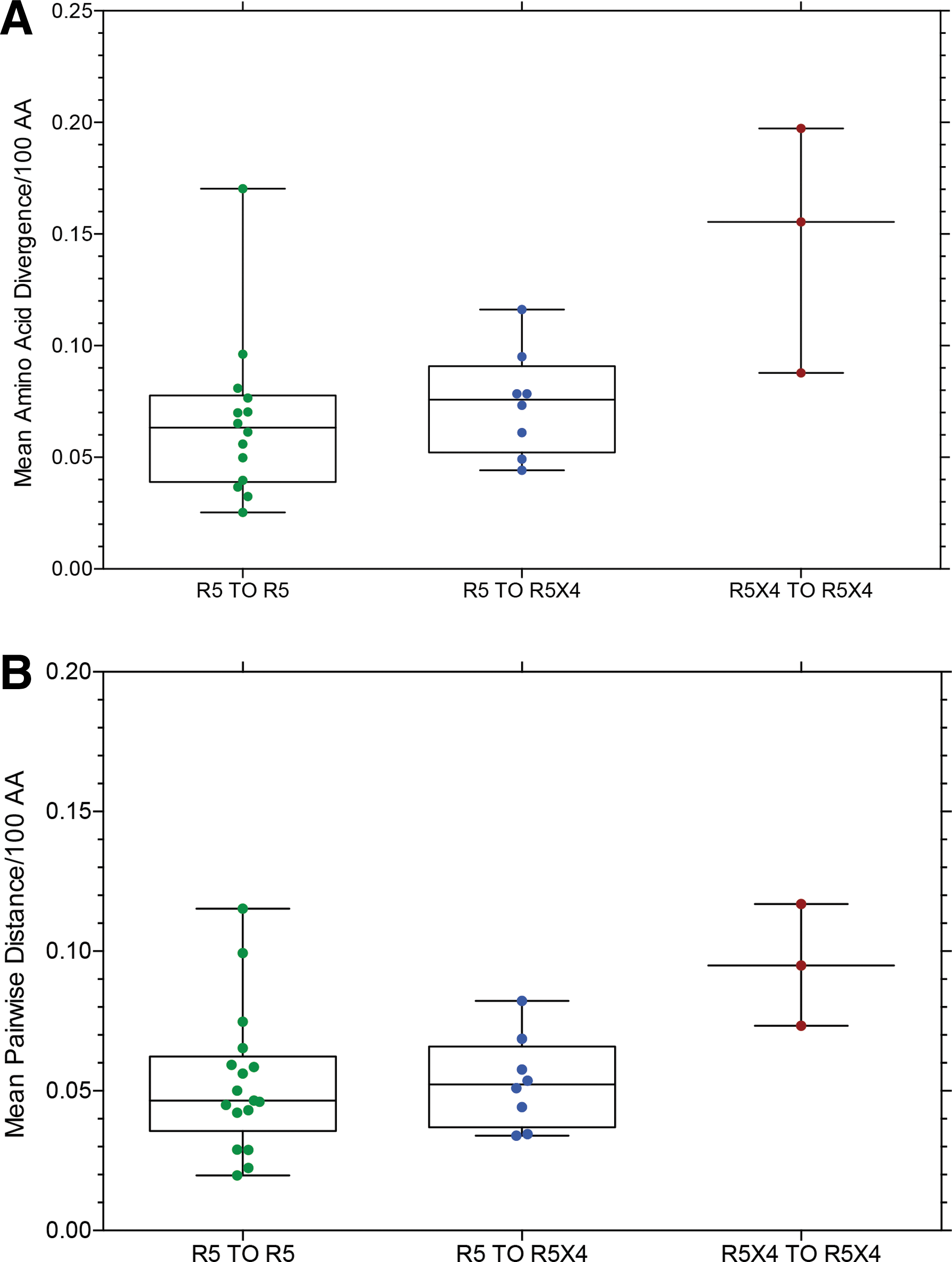

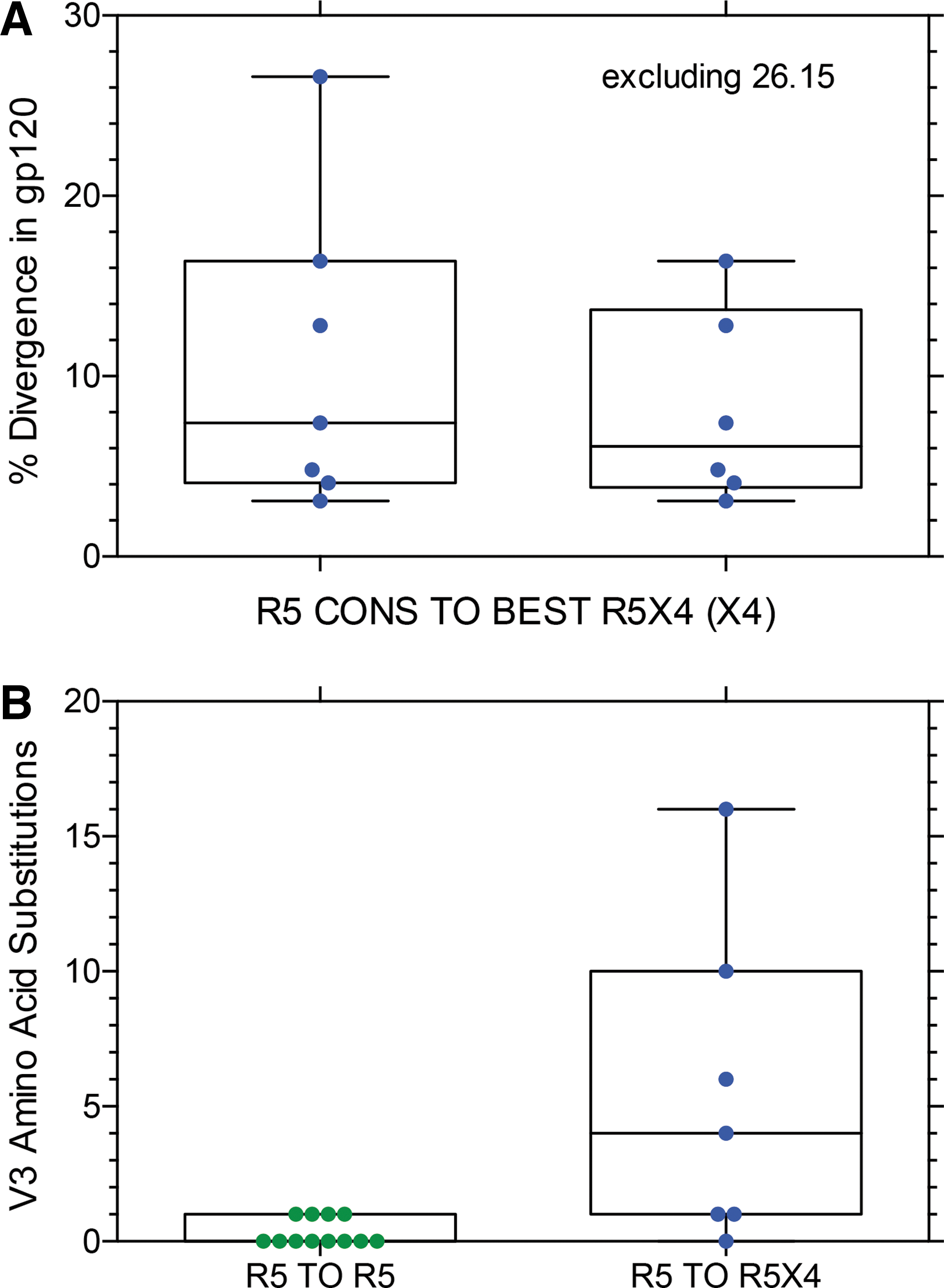
Figure 5 shows the percent divergence in gp120 amino acids from the consensus R5 sequence in sample 1 to the R5X4 (or X4 in two subjects) env clone that mediated the most efficient entry via CXCR4 in sample 2. Figure 5A shows the data with or without the highly divergent S26.15 sequence. If the outlier 26.15 (and two closely related sequences; see Fig. 9 below) is excluded, the median divergence from consensus R5 to the R5X4 variant that mediated the most efficient entry via CXCR4 4 months later is 6.1%, which is not significantly different than the median value (7.6%) for all R5 to R5X4 env clones shown in Figure 3A. Examination of all phylogenetic trees (Supplementary Figs. S1–S24) explains this somewhat surprising result. Although several subjects had substantial divergence between the first sample R5 consensus and the second sample R5X4 or X4 variant, other subjects had R5X4 sequences that were more closely related to the earlier R5 consensus than most contemporaneous R5 env sequences. Figure 5B compares divergence in the V3 sequence in the R5 to R5 control group to the divergence in the R5 to R5X4 switch group. This difference is highly significant as might be expected, given the importance of the V3 sequence in determining coreceptor use. However, genotypic predictors based on the V3 sequences performed poorly for these samples (data not shown), perhaps because of the recent (< 4 months) gain of entry via CXCR4.
Functional changes in envelope properties in coreceptor switch subjects versus NS controls
We evaluated the entry efficiency of all env clones capable of CCR5 use from subjects in the R5 to R5 NS group compared to the R5 to R5X4 coreceptor switch (S) group. The results are shown in Figure 6. Virus entry via CCR5 was significantly higher for the NS subjects compared to the S subjects (Fig. 6A) when results were pooled for both sample time points. The decreased entry via CCR5 was evident both before and after coreceptor switching (Fig. 6B), but the lower entry function in the preswitch samples no longer achieved significance. There were no consistent trends toward improved or declining entry efficiency during the interval between samples that distinguished NS controls from R5 to R5X4 S subjects (data not shown).
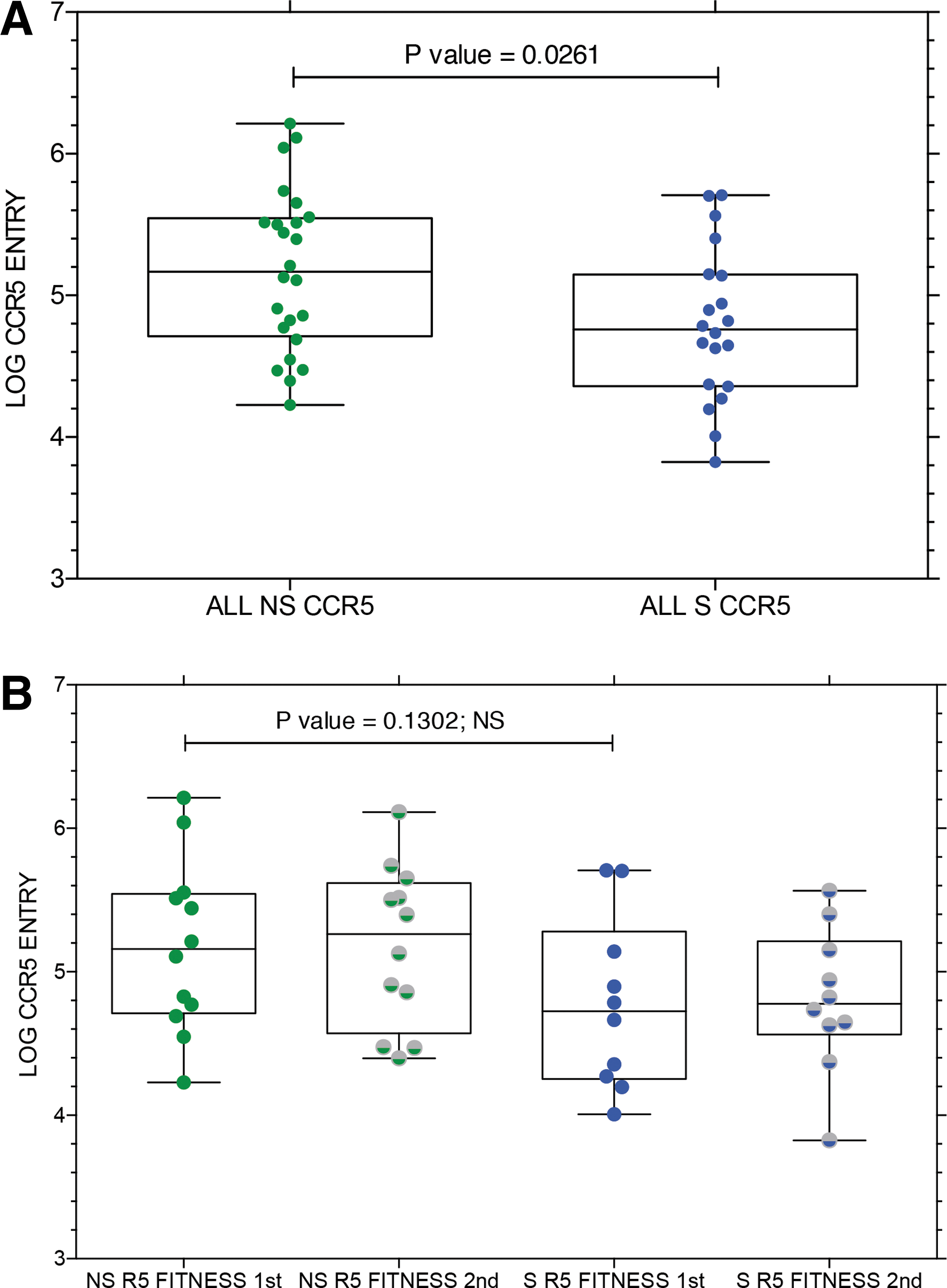
We also determined if there were any consistent changes in env sequence properties associated with coreceptor switching, and found no significant differences in potential N-linked glycosylation sites that distinguished env sequences from NS controls and sequences from S subjects (data not shown). However, V1/V2 regions were significantly longer in env sequences from R5 to R5X4 switching subjects (Fig. 7A) than control subjects when sequences from both samples were compared. When the change in V1/V2 length between the two sample times was compared (Fig. 7B), the NS controls tended to maintain the shorter V1/V2 length, while the S subjects showed a trend toward shorter V1/V2 length at the second sample point.

Many subjects in this study had prior analysis of viral replicative capacity 64 due to changes in protease (PR) and RT genes from their multidrug-resistant virus. Drug resistance mutations for most subjects are presented in Table S1. We used these data to determine if there was any correlation between reduced replication capacity (RC) and entry efficiency via CCR5, as suggested by a recent publication by Mohri et al. 65 We observed a trend in our sampling toward improved CCR5 entry as PR/RT RC declines, in agreement with Mohri et al., 65 but this trend was not statistically significant (p = .0603).
Clinical data from subjects that correlated with coreceptor switching
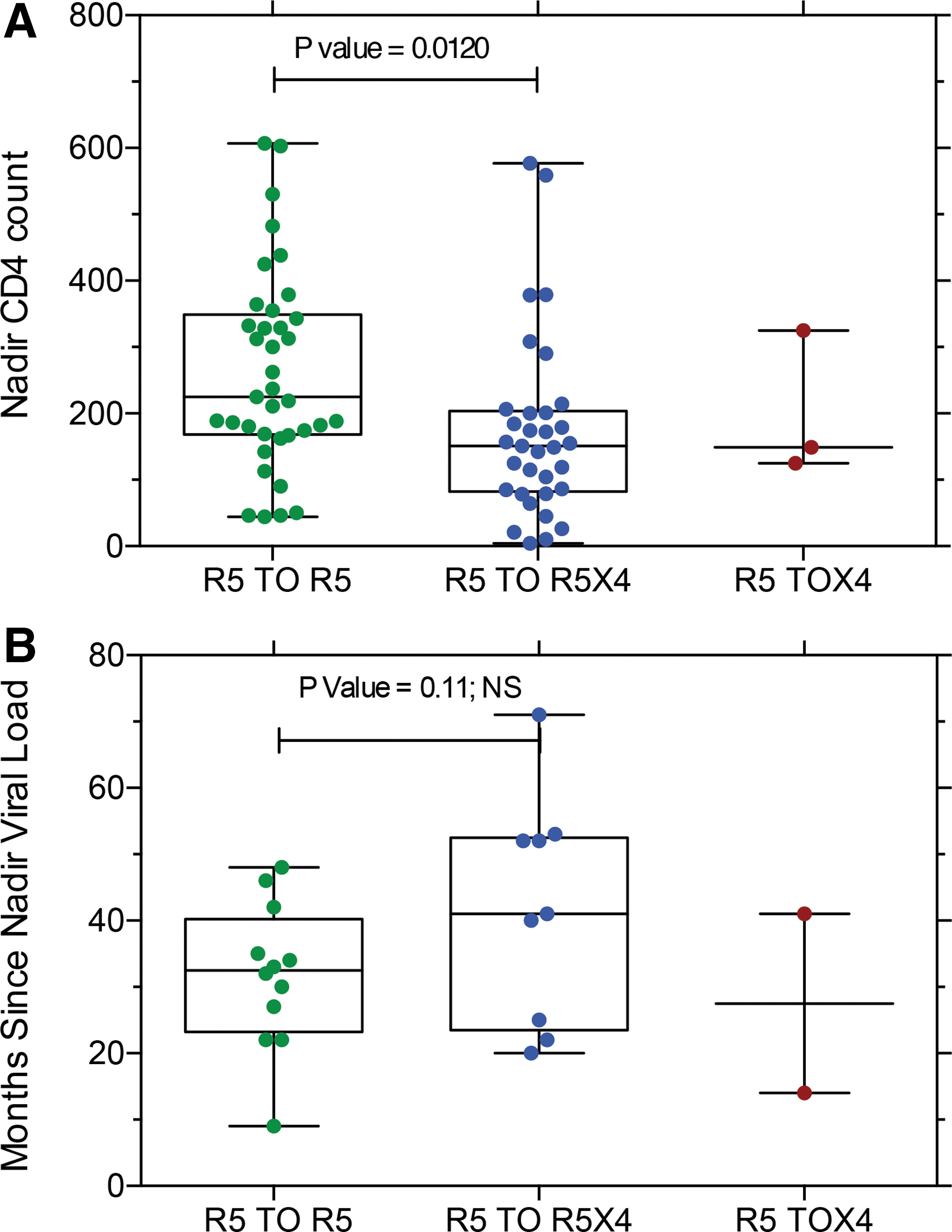
We evaluated two clinical parameters that might be associated with coreceptor switching, nadir CD4 T cell count 37 and the interval since the last nadir viral RNA load (Fig. 8). Lower nadir CD4 T cell counts were predictive of coreceptor switching (Fig. 8A) in agreement with prior results. 37 Subjects with coreceptor switching tended to have a longer interval since the nadir viral load, but the difference was not significant (Fig. 8B).
Extreme divergence or rescue of a latent virus from prior super-(or dual-) infection
During these studies, we noted two examples of env sequences at the second (or last) sample time point that were extremely divergent from any prior sequences. A phylogenetic tree of sequences from one subject with four sample time points is shown in Figure 9. Three closely related R5X4 env clones (26.2, 26.15, and 26.41, red shading) were 28% divergent from sequences from the prior sample time 104 days earlier (yellow shading), and share unique V3 regions. Clones 26.6 and 26.8 (red arrows) were recombinants that shared regions of close homology with both the minor and major strains. No other subject showed evidence of super-(or dual-) infection, and these highly divergent sequences were excluded from other data calculations.
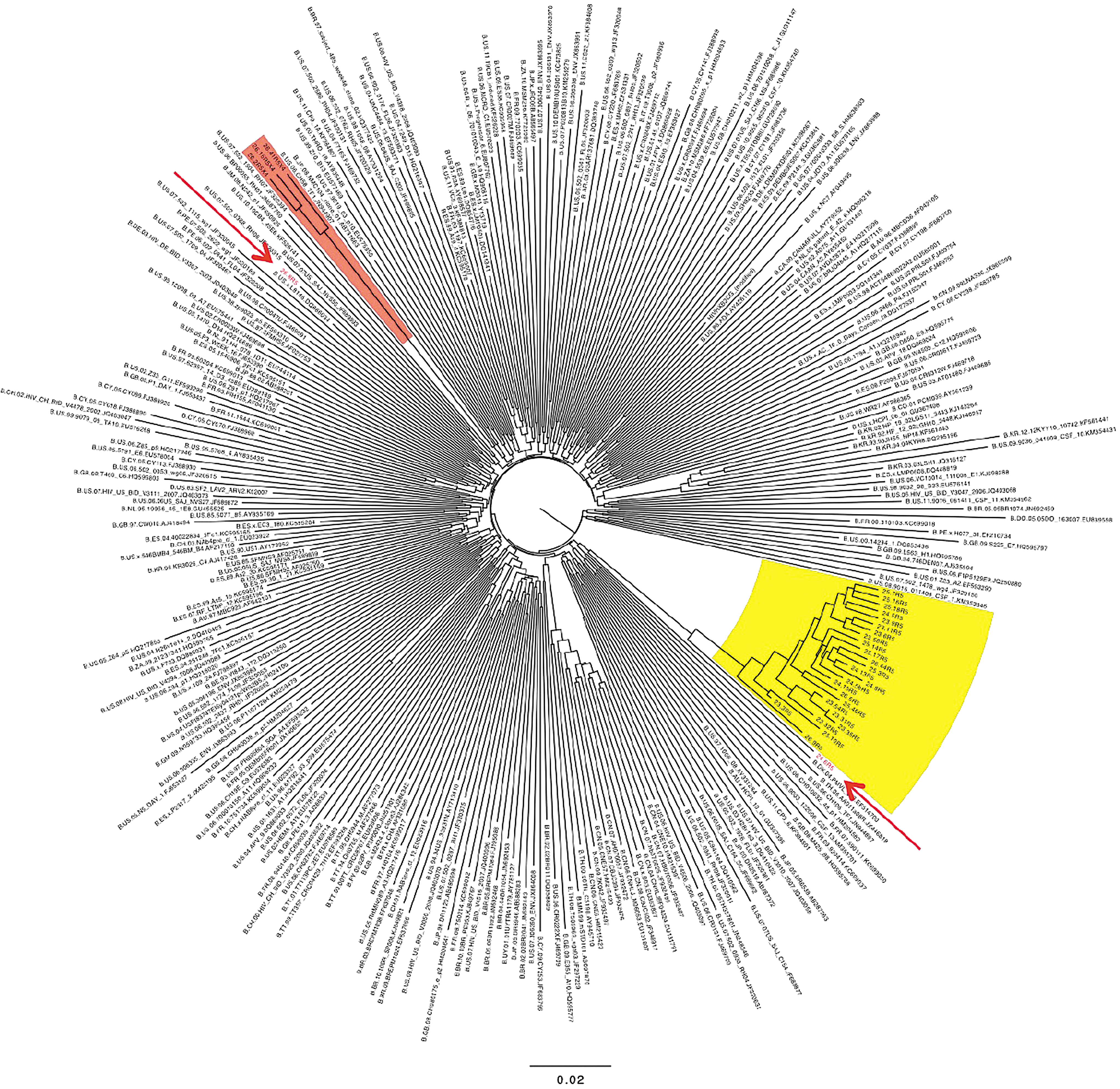
Neighbor-joining phylogenetic tree from dual/superinfected subject. The tree includes 30 env sequences from subject 3, and 102 and 290 randomly chosen subtype B sequences (see Materials and Methods section). Major and minor clades found in this individual are shown in yellow and red highlighted regions, respectively. Two recombinant env sequences containing regions derived from both the major and minor clades are in red type and indicated with arrows.
Discussion
Our results show that HIV-1 isolates from chronically infected, ART-experienced subjects with multiple drug-resistant mutations have both a great diversity of viruses (many with inferred diminished RC) and rapid turnover of virus populations from which variants with either enhanced entry function via CCR5 or gain of entry function via CXCR4 can be drawn. While we expected 66 –68 that env sequences from subjects with recent coreceptor switching would show greater divergence from the most recent common R5 ancestor than sequences from control subjects without coreceptor switching, this was not observed. Instead, the results in Figures 2 –5 show equivalent diversity and divergence for all env sequences independent of whether or not coreceptor switching had occurred between the sample intervals. The finding of equivalent diversity and divergence was sustained even when we limited analysis to the consensus R5 env sequences before coreceptor switching and the most robust R5X4 or X4 env sequences after switching (Fig. 5A). The only exception to this finding was the expected greater divergence in V3 sequences (Fig. 5B) associated with the R5 to R5X4 phenotypic switch. It should be emphasized that all sequence data were generated from env clones previously demonstrated to mediate virus entry via one or more coreceptors, so any defective env sequences that might be expected from plasma samples 69 had already been excluded from the sequence analysis.
The high diversity observed in this study was reminiscent of the recent report of Rothenberger et al., 70 where extensive diversity was found in rebound viruses after a short, treatment-interruption trial. Mean diversity ranged from 0.7% to 5.3% and maximum intrapatient diversity ranged from 2.2% to 9.0% in their study, and was evident by 2–3 weeks after cessation of ART. All of the subjects in our study were viremic at the time of both plasma samples (Table 1), and mean diversity ranged from 3% to greater than 20% (Fig. 3), reflecting the time of infection and/or the presence of dual- or superinfecting strains. 71 There have been relatively few longitudinal studies of envelope evolution during coreceptor switching where HIV-1 tropism was determined by functional assays rather than by genotypic predictors. Nonetheless, several recent studies yielded results that are similar to ours. Mild et al. 72 examined longitudinal V1–V3 env sequences from eight treatment-naive subjects and concluded that predicted X4 variants evolved at a higher rate than R5 populations. This conclusion is not supported by our results, where four subjects with coreceptor switching had more divergent R5X4 or X4 env sequences and the other four subjects had less divergent R5X4 env sequences than R5 sequences (excluding the dual- or superinfected subject). However, our subjects were treatment experienced and the added selection pressure of ART may have contributed to our results. In addition, we evaluated entire gp160 sequences, not just V1–V3 sequences. Sede et al. 73 examined C2–V3–C3 env sequences from 19 subjects with samples collected yearly and inferred coreceptor use with the Geno2pheno tool with a 10% false-positive cutoff. They observed considerable diversity in the short sequences from most subjects, and predicted X4 variants were interspersed among different R5 clades. Bunnik et al. 25 compared V3 sequences obtained by deep sequencing to phenotypic or genotypic assays for coreceptor use in longitudinal samples from eight individuals, and found X4-predicted V3 sequences as minor populations 3–6 months before phenotypic detection of X4-variants. As in our studies, there were major differences between subjects in both the diversity of R5 and X4 variants and the diversification between samples.
Three findings differentiated samples from those subjects with coreceptor switching compared to those with maintained R5 virus. Entry function of env clones via CCR5 was significantly lower in the coreceptor switch group (Fig. 6) before and after phenotypic switch from R5 to R5X4 virus. This observation supports the earlier hypothesis that loss of entry fitness is one factor leading to the emergence of R5X4 or X4 variants. 3,74 The V1–V2 length was longer in the env clones from the subjects with coreceptor switching (Fig. 7), although this trend was more evident in the sample before switching than in the subsequent sample after switching. Changes in V1–V2 sequences have been observed previously to impact coreceptor use, 8,9,75 and longer V1–V2 length has been associated with CXCR4 use in two prior studies. 76,77 V1–V2 length tends to increase with longer duration of infection. 63 However, longer V1–V2 regions may impair CD4 binding, 78 which may need to increase in HIV-1 envs with poor entry via both CCR5 and CXCR4. Nadir CD4 T cell counts were significantly lower in subjects with coreceptor switching than those without (Fig. 8) even though CD4 T cell counts at the time of sample collection were similar (Table 1), in agreement with the prior study 37 of these same subjects. This observation supports the paucity of available target cells as one factor favoring coreceptor switching 79 –81 if one assumes that CD4+, CXCR4+ target cells remain, while susceptible CD4+, CCR5+ target cells are depleted. 82 However, low nadir CD4+ T cell counts also correlate with immune dysfunction and poorer responses to ART, 43,83 so the direct contribution of lower target cell numbers to coreceptor switching is confounded by other potential selective forces.
Several observations made in this study are worth noting even though they did not achieve statistical significance. First, the number of potential N-linked glycosylation sites did not differentiate R5 and R5X4 or X4 env sequences in these subjects. Second, CCR5 entry fitness tended to increase as PR/RT RC decreased in agreement with Mohri et al. 65 Third, the duration since the last viral load nadir tended to be longer in those subjects with coreceptor switching, suggesting that a longer period of viremia as opposed to the current viral load might be important. These observations will require further studies to confirm. In addition, we searched the env sequences for evidence of recombination contributing to coreceptor switching. 84,85 Env clones from one subject infected from a single source and the individual with dual/superinfection showed evidence of recombination, and these clones were unique in that they shared a consensus V3 region with many R5 clones in the first instance and the majority strain in the second.
The low replicative and entry fitness of many variants may explain the rapid turnover of env genotypes. This low fitness landscape may present an opportunity for many divergent viruses to emerge, but a distinct fitness winner was rarely observed. In only one subject did we observe duplicate env sequences at one sample point, and the high level of divergence meant that few closely related sequences were observed at both sample time points. The two subjects with the longest duration of diagnosis and R5X4 viruses at both sample time points had the highest PR/RT RC scores but still had rapid turnover of env sequences and high diversity, suggesting that PR/RT replication fitness alone does not lead to a dominant HIV-1 env sequence.
Virus evolution is driven by a constant interplay between mutation and recombination of the viral genome and selective pressure exerted by many factors, including the host immune system, ART, and the need to maintain replication and entry fitness. 3,48,72,86 –91 In the setting of chronic infection with poor or intermittent responses to ART, diminished PR/RT RC, poorer entry fitness via CCR5 (for those subjects with coreceptor switching), and diminished immune responses were indicated by lower nadir CD4+ T cell counts, perhaps it should not be surprising that coreceptor switching is more prevalent at equivalent CD4+ T cell levels than in treatment-naive subjects. 32,37
Footnotes
Acknowledgments
We thank Suqin Cai, Jonathan Toma, and Jeannette Whitcomb for assistance with sequence analysis of the subject with dual/superinfection and Yolanda Lie for support with project management. This work was supported by NIH grants AI104313 to DEM and AI52745 to SGD. Data analysis reported in this publication was also supported by NIAID, NCI, NIMH, NIDA, NICHD, NHLBI, NIA, NIGMS, NIDDK of the National Institutes of Health under CFAR award number AI027757 to the University of Washington and AI27763 to the University of California, San Francisco. This publication was also supported by the CFAR Network of Integrated Systems (R24 AI067039) and the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI grant number UL1 TR000004. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author Disclosure Statement
J.D.R. and E.D.A. are employees of Monogram Biosciences, Laboratory Corporation of America Holdings (LH), and shareholders of LH. The other authors report no conflicts of interest exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.