
Abstract
Human foamy virus (HFV) is a complex and unique retrovirus with the longest genomes among retroviruses that are used as vectors for gene therapy. Long non-coding RNAs (lncRNAs) are regarded as key regulators that are involved in diverse biological processes during viral infection. However, the role of lncRNAs in HFV infection remains unknown. In this study, we utilized next-generation sequencing to first characterize lncRNAs in 293T cells after HFV infection, evaluating length distribution, exon number distribution, volcano picture, and lncRNA class distribution. We identified 11,336 lncRNAs (4,729 upregulated lncRNAs and 6,588 downregulated lncRNAs) and 61,367 mRNAs (30,133 upregulated mRNAs and 31,220 downregulated mRNAs), which were differentially expressed in the HFV-infected 293T cells. Subsequently, six differentially expressed lncRNAs characterized using RNA-seq were confirmed by quantitative real-time polymerase chain reaction assays. Interestingly, Gene Ontology (GO)/Gene Ontology Tree Machine (GOTM) and Kyoto Encyclopedia of Gene and Genomes (KEGG) pathway analyses indicated that positive regulation of interleukin 8 (IL8) production and cytokine–cytokine receptor interaction might be involved in the functional enrichment of lncRNAs. Moreover, cis-acting and trans-acting regulatory networks show that NR_028036 may target the fas gene in a cis-acting manner and that ENST00000354838 may target the IL18 gene in a trans-acting manner. Overall, these results not only provide novel insights into the relationship between HFV and lncRNAs in the host response to infection but also have implications for the future wider application of HFV as a vector.
Introduction
H
Long non-coding RNAs (lncRNAs) are considered as RNA transcripts with no evident protein-coding potential and are longer than 200 nucleotides in length. 13 Most lncRNAs are enriched in the cell nucleus 14 and usually have two to four exons and a polyA tail. 13 It has been reported that only ∼2% of RNAs can be translated into proteins and that the remaining 98% are non-coding RNAs. 15 Currently, more than 100,000 lncRNAs have been found in humans alone. 16 Since lncRNAs were first described in 2002, 17 they have been implicated in transcription modulation, signal transduction, molecular scaffolds, nuclear organization, and epigenetic regulation. 18 –22
Recent studies have demonstrated some relationships between host lncRNAs and viral infections. After establishment of Epstein Barr virus latent infection, host lncRNA increased and, subsequently, interacted with a number of cellular proteins. 23 lncRNA NeST can influence susceptibility to Theiler's murine encephalomyelitis virus infection. 24 During EV71 or CA16 infection, lncRNAs are differentially expressed in human rhabdomyosarcoma cells (RD cells). 25,26 lncRNA NEAT1 is upregulated in HIV-1 infection and modulates HIV-1 posttranscriptional expression. 27 lncRNA noncoding repressor of Nuclear Factor of Activated T cells (NRON) is oppositely regulated by HIV-1 early and late viral proteins, and NRON knockdown enhances HIV-1 replication in a Nuclear Factor of Activated T cells-dependent manner. 28 However, the dynamics of lncRNA expression remain unclear with respect to the host response to HFV infection. In this study, we performed RNA-seq analysis on HFV-infected and -uninfected 293T cells to characterize lncRNA expression.
Materials and Methods
Cell culture and virus
293T cells (human embryonic kidney cells) and HT1080 cells (human fibrosarcoma cells) were purchased from the American Type Culture Collection. 293T cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS; HyClone) at 37°C. HT1080 cells were cultured in minimum essential medium supplemented with 10% FBS (HyClone) at 37°C. HFV was produced by transfecting 293T cells with provirus plasmids pHSRV13 29 (German Cancer Research Center, Heidelberg, Germany). After 48 h at 37°C, virus was released by freezing and thawing transfected cells and the culture medium for three cycles.
RNA extraction and quantitative real-time polymerase chain reaction
Trizol reagent (Invitrogen) was used to extract total RNA from 293T cells or HT1080 cells according to the manufacturer's instructions. The concentration and purity of RNA was tested by an ultraviolet spectrophotometer (Eppendorf). The value of OD260/OD280 ratio can be used as a reference of RNA purity. Our data of OD260/OD280 ratios were in the range from 1.9 to 2.1, which suggested that the total RNAs can be used for the succeeding experiment. Then, Reverse Transcription kit (Thermo) was used to transcribe 2 μg RNA into 20 μl cDNA, in line with the protocol. Differential expression of lncRNAs was validated by quantitative real-time polymerase chain reaction (qRT-PCR) (Bio-Rad iQ5) by using SYBR green (Toyobo), and Actin was used as a control gene. Primers of six selected lncRNAs and Actin were designed on the website of Intergated DNA Technologies (
lncRNA, long non-coding RNA.
Secondary structure predictions of lncRNAs
Secondary structures of lncRNAs were predicted by using the RNAfold web server (
RNA-seq and data analysis
HFV-infected and -uninfected 293T cells were sent to Genenery Bio-Tech for sequencing. RNA-seq was performed by using the Illumina Hiseq2500 platform, and Trim Galore software was used to remove low-quality sequences. FastQC software was used for quality control analysis. Tophat software was used to compare pretreatment sequences and whole-genome reference sequences. Cufflinks software was used to reconstruct transcripts. Predictions of new lncRNAs were made by using HMMer+Pfam/CPC/PhyloCSF/CPAT theories. Differential expression of lncRNA was analyzed by using fragments per kilobase of transcript per million fragments mapped (FPKM) calculating metrics.
Gene Ontology/Gene Ontology Tree Machine and Kyoto Encyclopedia of Gene and Genomes pathway analyses
Gene Ontology (GO) 30 analysis was used to classify and annotate whole genome/transcripts and differentially expressed genes/transcripts. Specifically, the hypergeometric distribution test was used to calculate whether differentially expressed genes/transcripts existed (enrichment p-value), and then the Benjamini–Hochberg multiple test was applied to correct the p-value to obtain the false discovery rate. Gene Ontology Tree Machine (GOTM) 31 generated GOTrees based on a GO enrichment p-value <0.05. Kyoto Encyclopedia of Gene and Genomes (KEGG) 32 pathway analysis is similar to GO analysis, but KEGG analysis uses the KEGG database.
Prediction of cis-acting and trans-acting regulatory networks
Cis-acting regulation refers to the direct regulation of protein genes by lncRNAs. Protein genes near sense lncRNAs, antisense lncRNAs, intronic lncRNAs, enhancer lncRNAs, and bidrectional lncRNAs were considered potential target genes in cis-acting regulation. Using RepeatMasker software, SINE type was tested between lncRNA and mRNA to obtain cis-acting regulatory networks. Trans-acting regulation refers to the indirect regulation of protein genes by lncRNAs through sequence complementation due to the distance between each factor. Studies have shown that lncRNAs can interact with the 3′UTR of protein genes through an ALU repeat sequence for transcription regulation. Therefore, the RNAplex and Risearch programs were used to predict trans-acting genes.
Results
Characterization of lncRNAs during HFV infection using RNA-seq
To determine whether host lncRNAs change after HFV infection of 293T cells, genome-wide RNA-seq was used to compare lncRNA expression in infected and uninfected cells. Length distribution, exon number distribution, volcano picture, and lncRNA class distribution were characterized. As shown in Figure 1, the majority of lncRNAs are less than 10 kb in length (Fig. 1A), and most lncRNAs comprise 20 or fewer exons (Fig. 1B).
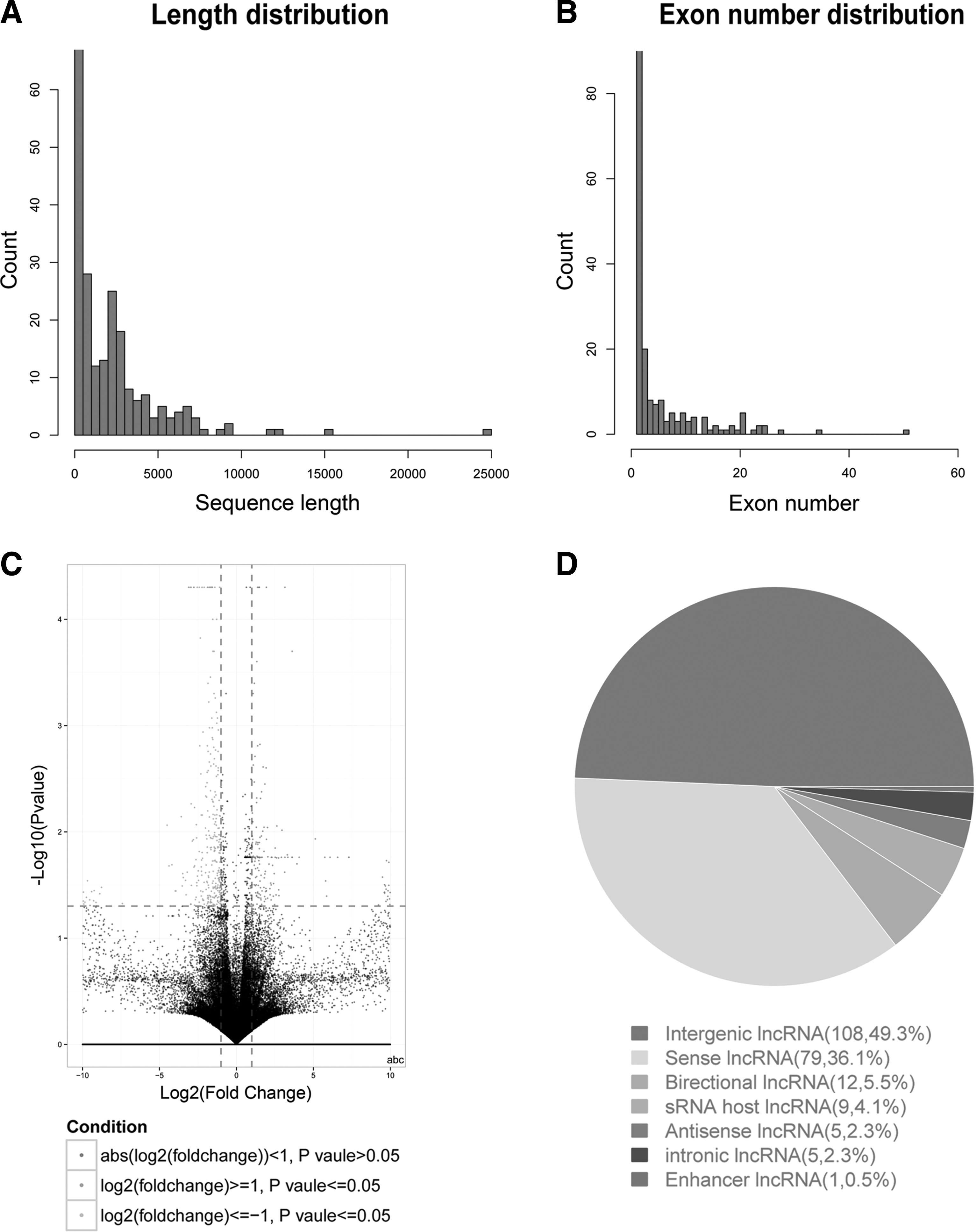
Basic information of RNA-seq.
The expression of lncRNA transcripts was calculated by FPKM. FPKM defines every 1 kb length of pairing sequences as the standard of transcript expression. Transcript length and number of pairing sequences are used as the unifying values of expression. The Cuffdiff program was used to screen differential expression of transcripts in samples of 293T cells infected or uninfected with HFV. The levels of difference between the two groups were reported by the volcano picture (Fig. 1C).
Based on the relative position of an lncRNA and a protein gene transcript, lncRNAs were classified into seven groups: sense lncRNAs, antisense lncRNAs, intronic lncRNAs, enhancer lncRNAs, sRNA host lncRNAs, bidirectional lncRNAs, and intergenic lncRNAs. The direction of sense lncRNA is the same as that of the protein gene transcript, and both exons overlap. The direction of antisense lncRNA is opposite to that of the protein gene transcript, but both exons also overlap. Intronic lncRNAs are located in the introns of protein gene transcripts. Enhancer lncRNAs overlap with enhancers of protein gene transcripts, and enhancer lncRNAs are located in protein gene transcripts or within 10 kb upstream or downstream of a protein gene transcript. sRNA host lncRNAs overlap with miRNAs. The distance between a bidirectional lncRNA and the initial position of a protein gene transcript is 1 kb, and the lncRNA and protein gene have opposite directions. Intergenic lncRNAs are located in the intervals between protein genes. The distribution of the lncRNA classes, as determined by RNA-seq, is 36.1% sense lncRNAs, 2.3% antisense lncRNAs, 2.3% intronic lncRNAs, 0.5% enhancer lncRNAs, 4.1% sRNA host lncRNAs, 5.5% bidirectional lncRNAs, and 49.3% intergenic lncRNAs (Fig. 1D). These RNA-seq results broaden our general understanding of lncRNAs during HFV infection.
Significant differential expression of lncRNAs and mRNAs in HFV-infected 293T cells
Because lncRNAs participate in various aspects of gene expression affecting normal host biological functions, it is not surprising that dysregulation of lncRNAs is involved in viral infections. Using RNA-seq, we compared changes in lncRNA and mRNA expression between 293T cells infected with HFV and uninfected cells. Significant differential expression of lncRNAs or mRNAs was indicated by a p-value <0.05. We identified 11,336 lncRNAs, including 4,729 upregulated lncRNAs and 6,588 downregulated lncRNAs, which showed significant differential expression in the HFV-infected group compared with the HIV-uninfected group. Then, we further refined the results and found that 958 upregulated lncRNAs are log2 ≥ 2 and 1,168 downregulated lncRNAs are log2 ≤ −2. These significantly differentially expressed lncRNAs are found in several professional lncRNA databases: Ensemble (
Then, we randomly chose several significantly differentially expressed lncRNAs and mRNAs to construct heat maps. The distributions of the ratios of lncRNAs (Fig. 2A) and mRNAs (Fig. 2B) between HFV-infected and -uninfected 293T cells are characterized by different colors in each figure. Differences are shown on a scale from 2 to −2.
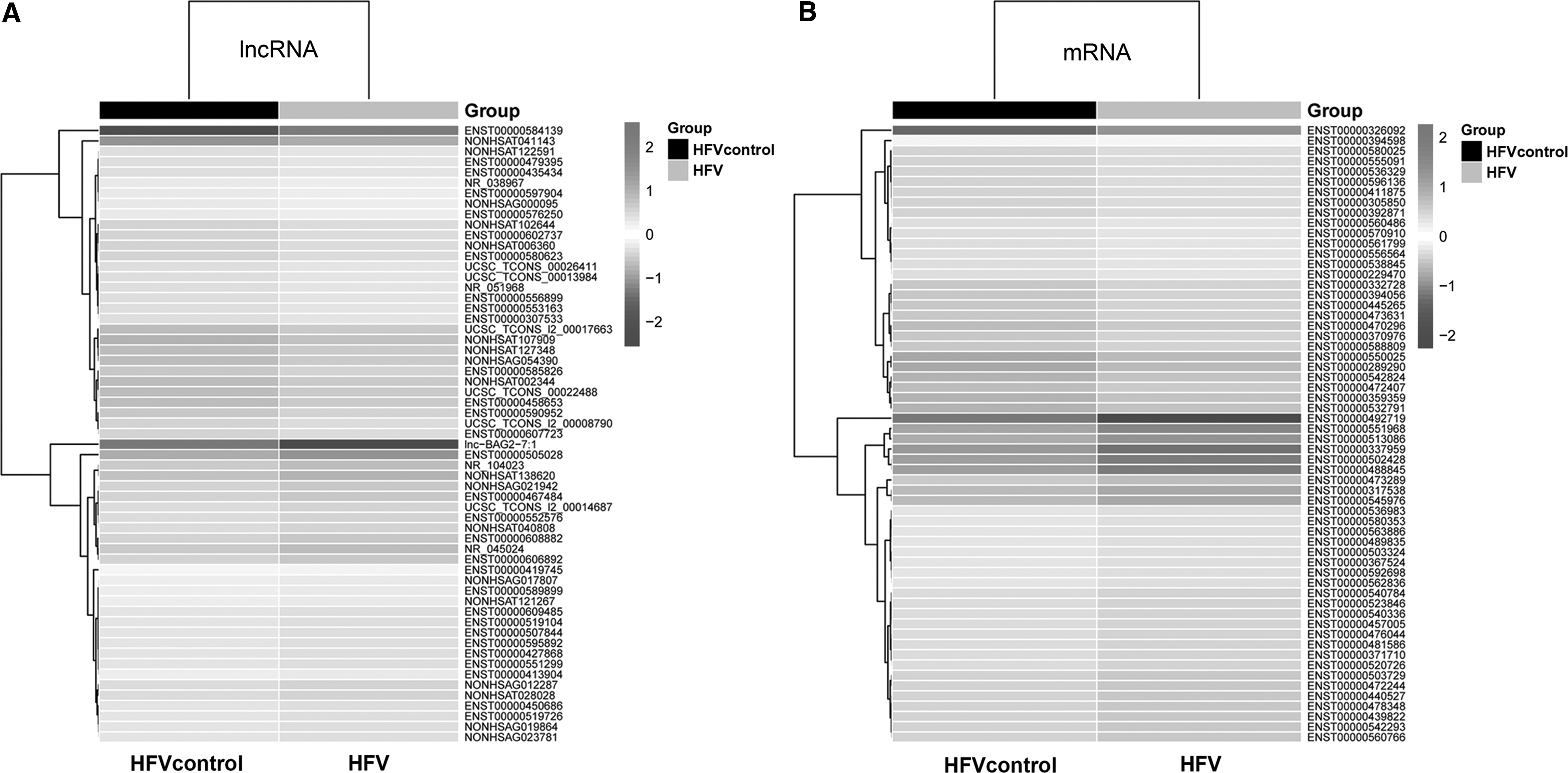
Heat maps of significant differentially expressed lncRNAs and mRNAs.
Differentially expressed lncRNA validation and secondary structure prediction
To validate the accuracy of RNA-seq, we performed qRT-PCR assays on six selected lncRNAs. The heat map of these lncRNAs was made by using RNA-seq data (Fig. 3A). Similar results were obtained from the qRT-PCR assay, in which two lncRNAs (NONHSAG000101 and ENSG00000204054) were upregulated and four lncRNAs (ENSG00000227254, ENSG00000247095, NONHSAT135354, and lnc-RP5-1086D14.3.1–1:1) were downregulated in HFV-infected 293T cells (Fig. 3B) and HT1080 cells (Fig. 3C). In brief, these data demonstrate the accuracy of the RNA-seq results.
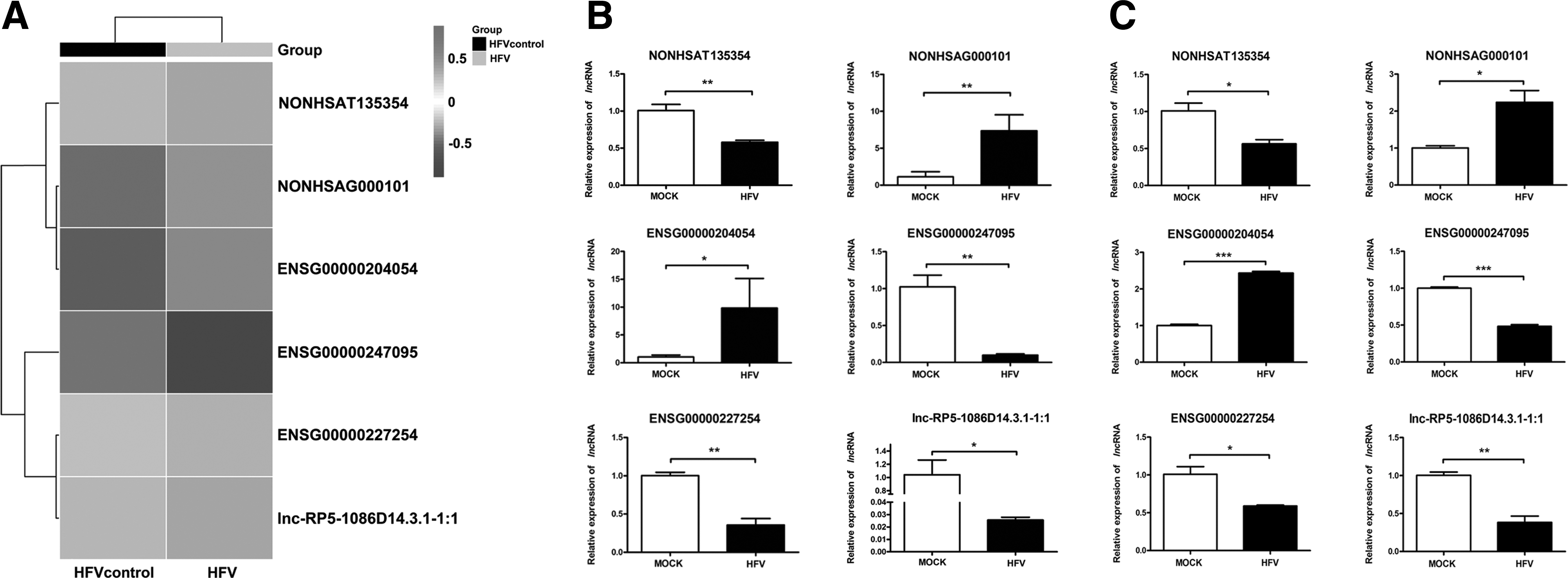
Validating differential expression of six selected lncRNAs in RNA-seq.
Bioinformatics analysis indicated that the structural conformation of lncRNA plays a crucial role in its biological effect, especially with respect to its secondary structure. 33 Structural diversity allows lncRNAs to perform functions that influence splicing, transcription, translation, and localization. 34 Consequently, the prediction of lncRNA secondary structure can provide valuable information regarding the versatility of lncRNAs in various cellular processes and lncRNA function. 35 Currently, multiple sequence alignments and MFE structures are two types of universally accepted ways to represent RNA secondary structure. 34 MFE structures were obtained by using a loop-based energy model and the dynamic programming algorithm. 36 With the help of the RNAfold Web Server, we used MFE structures to predict the secondary structure of six lncRNAs that have been validated (Fig. 4). The picture visualizes the stem-loop structures of lncRNAs, and their complexity can be judged based on the number of stem-loop structures. Moreover, we can estimate the base pairing potential of each lncRNA based on the number. One indicates the highest base pairing potential, and zero indicates the lowest base pairing potential.
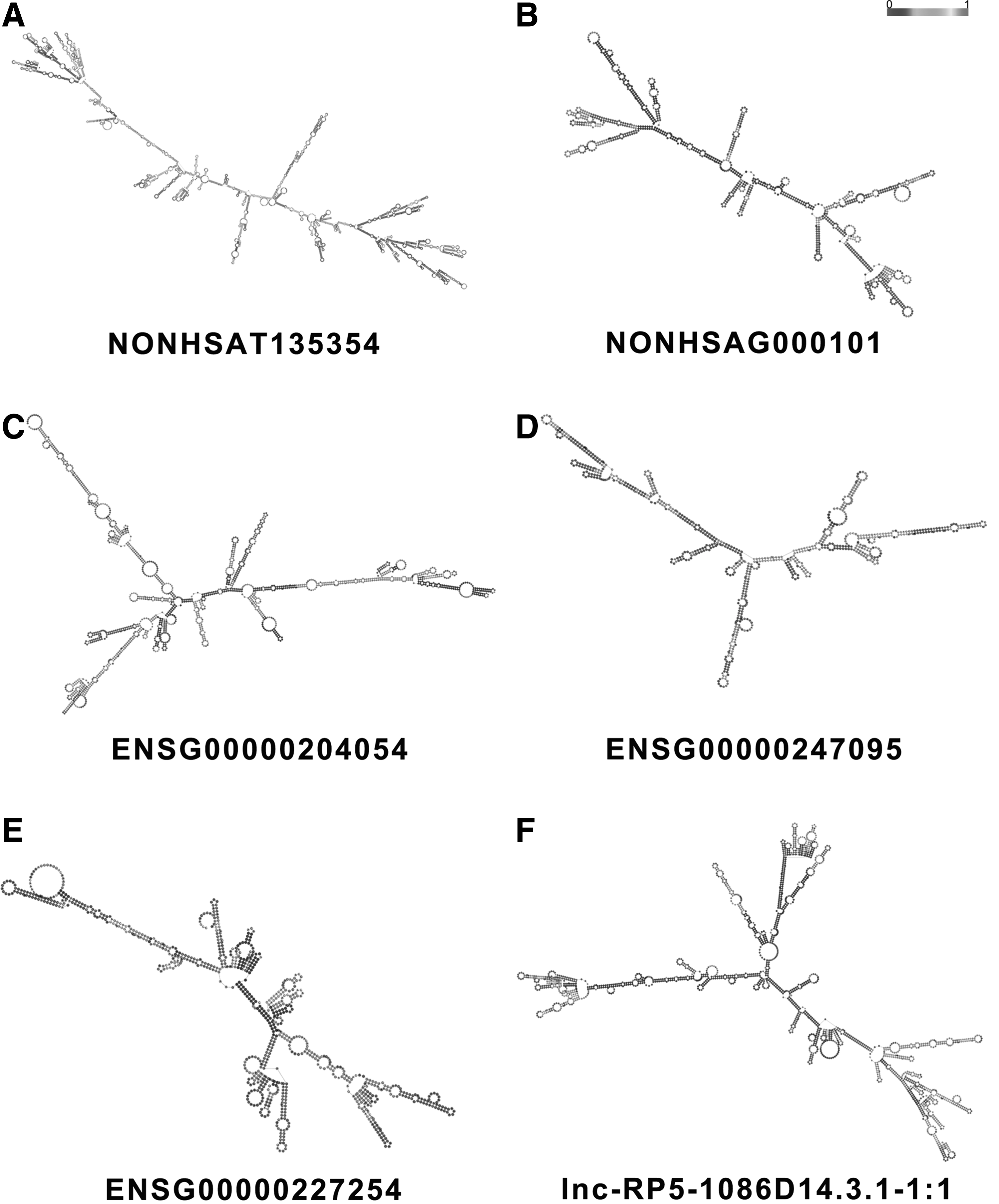
Secondary structure predictions of lncRNAs. MFE structures were used to predict the secondary structure of lncRNAs, and a scale from 0 to 1 represented base pairing possibility.
Functional enrichment analysis of differentially expressed genes during HFV infection
We used GO 30 /GOTM 31 and KEGG 32 pathway analyses to determine lncRNA function from RNA-seq data. GO pathway analysis is a useful tool for constructing and annotating widespread biological processes, cellular components, and molecular functions. The significant GO enrichment categories were defined as those with a p-value <0.05 between groups of 293T cells infected or uninfected with HFV. GO analysis showed three types of gene function enrichment: biological process, cellular component, and molecular function enrichment (Fig. 5A, B). In particular, the functions of differentially expressed genes are highly enriched in response to decreased oxygen levels and intracellular and protein binding in these three enrichment types (Fig. 5A). Notably, negative regulation of the homeostatic process and intracellular and N-acylmannosamine kinase activity are the three most significantly enriched functions in differentially expressed lncRNAs (Fig. 5B). Additional GO analysis may reveal enrichment of other functions.
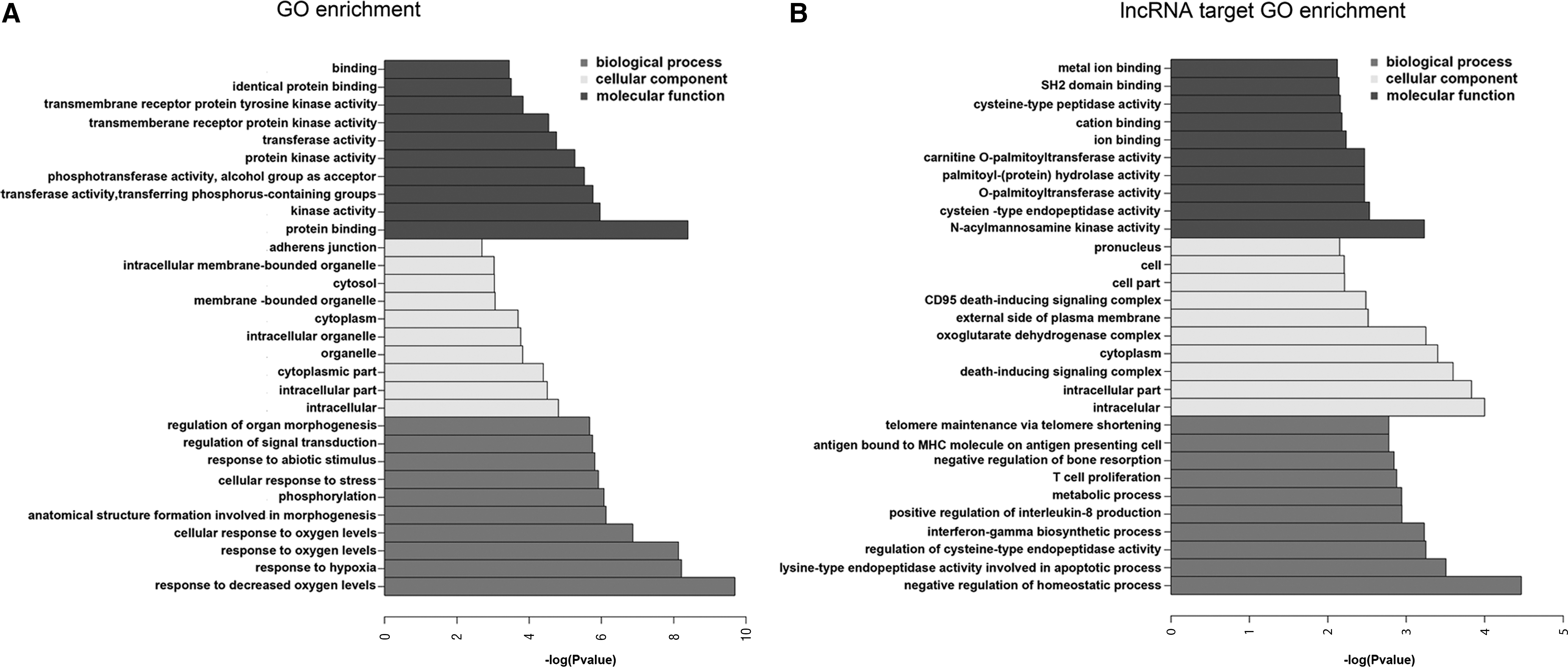
GO analysis and GOTM.
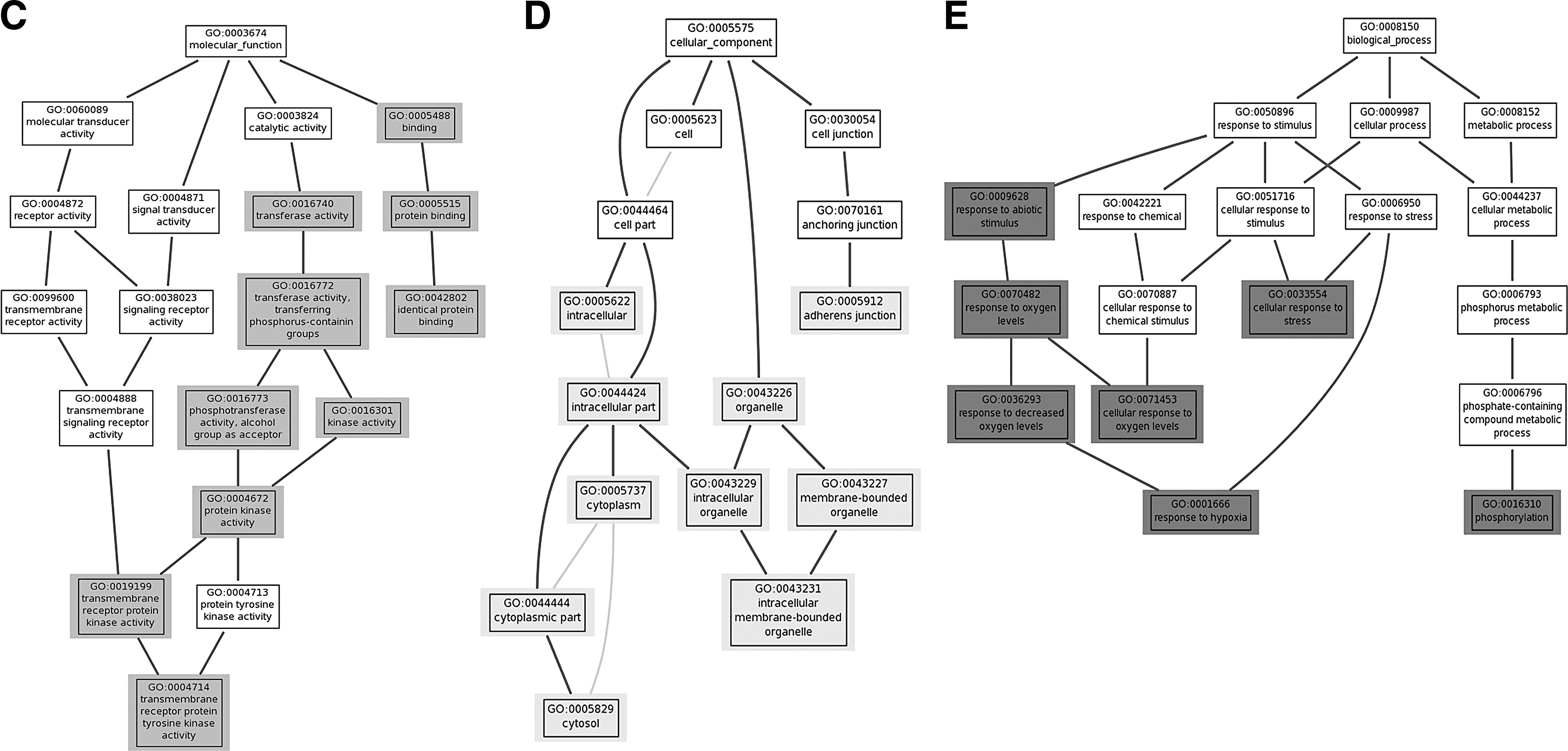
GOTM generated three types of GOTrees based on a GO enrichment p-value <0.05 (Fig. 5C–E). Figure 5C represents molecular functions. Figure 5D represents cellular components. Figure 5E represents biological processes. Shaded rectangles represent significant GO enrichment of GOTrees. Each rectangle contains a GO number that corresponds to a specific function.
KEGG pathway analysis was used to predict biological pathways associated with functions of differentially expressed genes. The HIF-1 signaling pathway is the most correlated pathway in KEGG enrichment (Fig. 6A). Similarly, the pathways of cancer, small cell lung cancer, focal adhesion, and others are also involved (Fig. 6A). Some of these pathways are associated with immunity, including the T cell receptor signaling pathway, TNF signaling pathway, and chemokine signaling pathway (Fig. 6A). Interestingly, cancer pathways are also involved in lncRNAs targeted by KEGG enrichment (Fig. 6B). African trypanosomiasis, taurine and hypotaurine metabolism, and basal transcription factors are enriched in lncRNAs targeted by KEGG enrichment (Fig. 6B). In addition, some pathways show no significant difference, including prion disease, rheumatoid arthritis, apoptosis, and others (Fig. 6B).
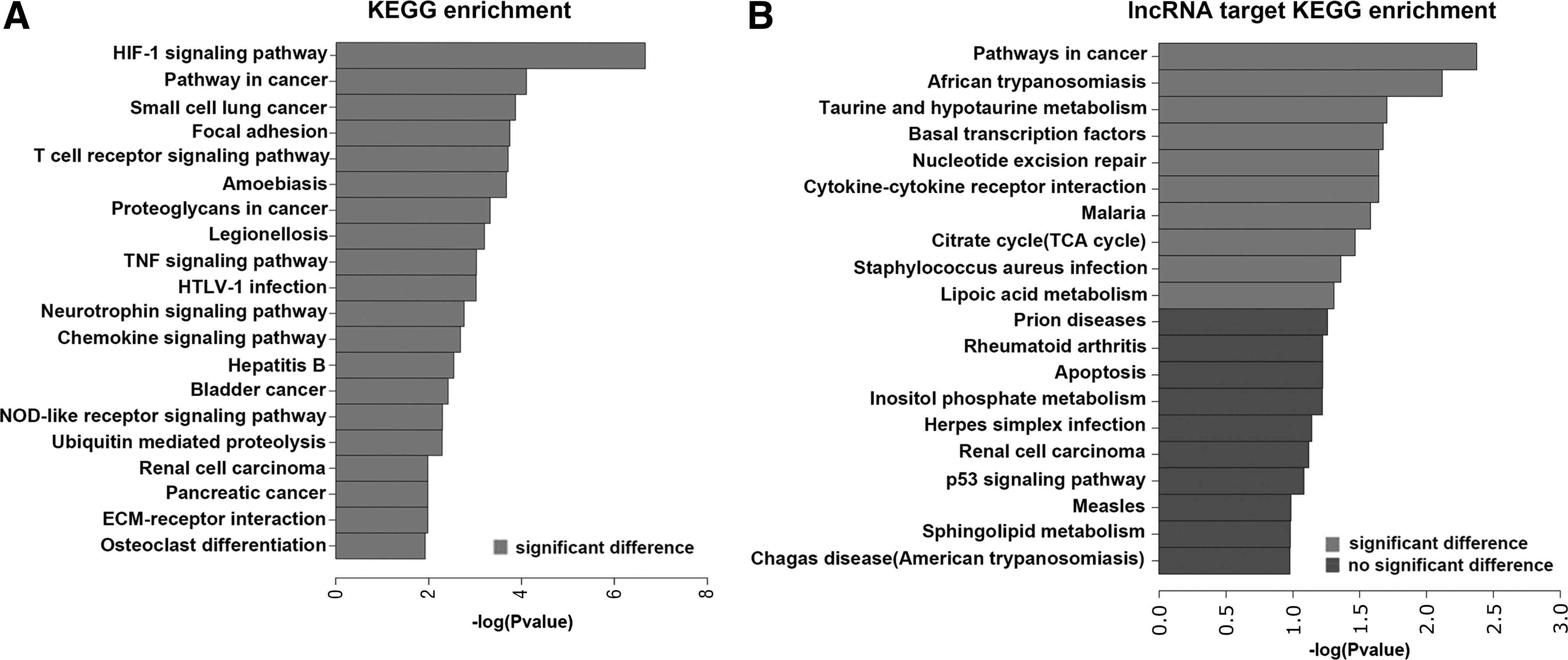
KEGG pathway analysis.
Cis-acting and trans-acting regulatory networks
It is widely acknowledged that no genes have functions in isolation; rather, they must interact with other genes in complex regulatory networks. 37 Much of the research on lncRNA regulation has illustrated that some lncRNAs regulate the expression of overlapping or nearby genes by cis-acting networks, including lncRNA Xist, H19, and Airn. 38 –40 Here, we identified 409 differentially expressed lncRNAs that target related protein genes in cis-acting manners. Some specific correlations are shown in Figure 7A. Previous studies have also shown that lncRNAs can regulate protein expression in trans-acting manners. For example, lncRNA HOTAIR was shown to remotely regulate coding transcripts from the HOXD cluster in a trans-acting manner. 41 Similarly, 637 differentially expressed lncRNAs were found to target related protein genes in a trans-acting manner. Two differentially expressed lncRNAs are shown as examples in Figure 7B. In addition, we found that two different lncRNAs might target the same protein genes. Thus, these results indicate a useful direction for the study of lncRNAs in specific gene relationships during HFV infection.
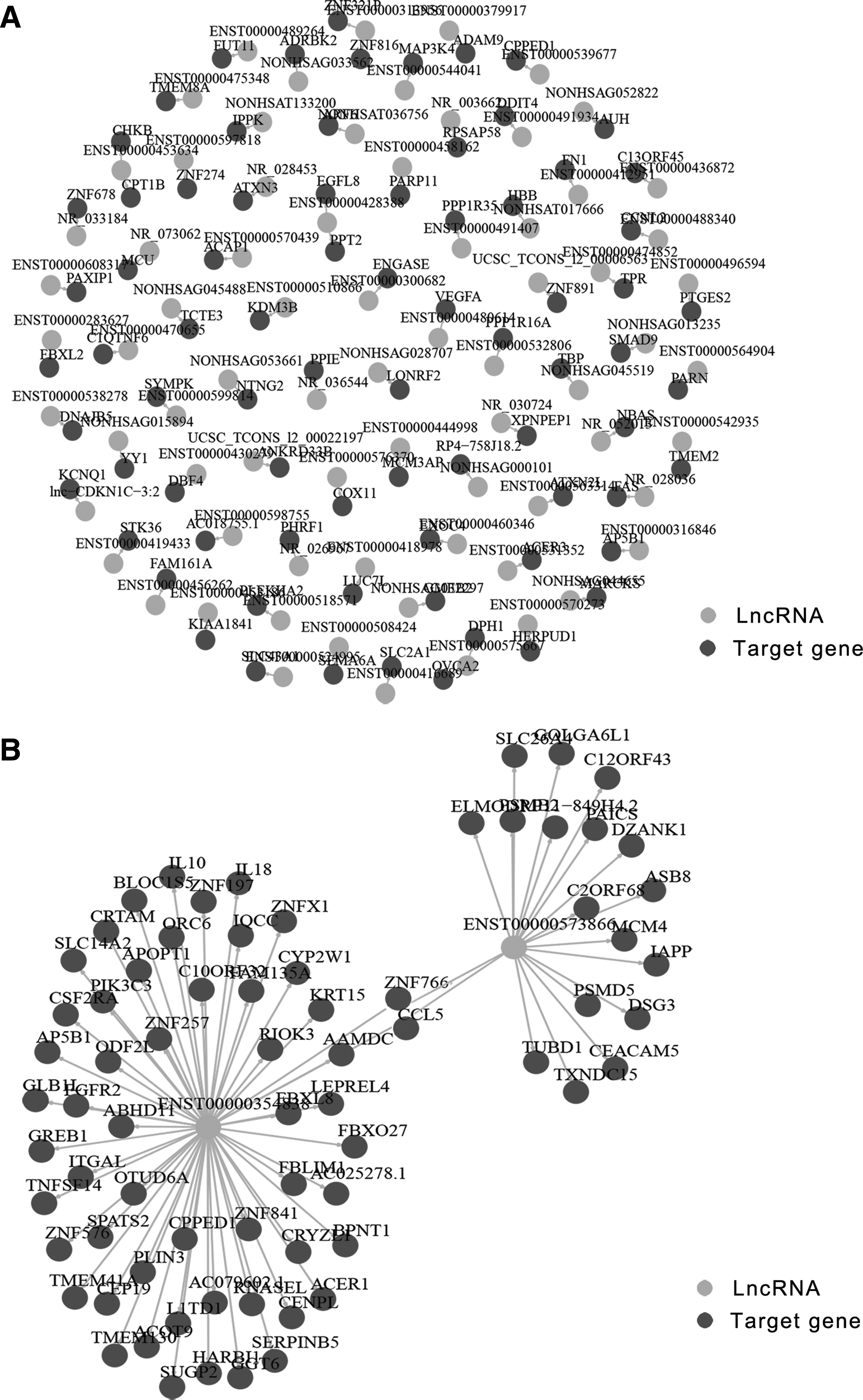
lncRNA cis-acting and trans-acting regulatory network.
Discussion
In this study, we found that lncRNAs and mRNAs were dysregulated in HFV-infected 293T cells. We identified 11,336 lncRNAs and 61,367 mRNAs as significantly differentially expressed by using next-generation sequencing. GO/GOTM and KEGG pathway analyses indicated several potential biological effects and regulatory mechanisms associated with these lncRNAs, which could be of use in further investigations. Furthermore, cis-acting and trans-acting regulatory networks provided more information on lncRNA-targeting genes, opening avenues for further research.
Three types of lncRNAs are currently recognized: antisense lncRNAs, intergenic (lincRNAs), and intronic lncRNAs. 42 There are ∼20,000 transcripts of these three lncRNAs described in current publications. 18 In our study, most of the lncRNAs were antisense lncRNAs or lincRNAs, which is consistent with previous RNA-seq investigations. For example, the GENCODE project revealed that there are 3,214 antisense lncRNAs and 5,058 lincRNAs but only 378 sense lncRNAs and 930 processes transcripts in the lncRNAs data. 43 Yin et al. reported that antisense lncRNAs and lincRNAs constitute a large part of all differentially expressed lncRNAs in EV71-infected RD cells. 25 Similar results were found by Shi et al., who showed that differentially expressed lncRNAs are almost all lincRNAs in CA16-infected RD cells. 26 The antisense form of lncRNA has a higher stability with respect to half-life, 44 and the functions of antisense lncRNAs are widespread in different tissues and cells. Moreover, previous studies of lncRNAs and HIV interactions showed that HIV can encode an antisense lncRNA that modulates HIV-1 gene expression through epigenetic alteration of the viral promoter. 45 As for lincRNAs, current publications show that nearly 3,300 lincRNAs analyzed by chromatin-state maps are involved in diverse biological processes, including cell-cycle regulation and immune surveillance. 46 Host lincRNAs may also be involved in viral infections. For instance, lincRNA VIN was reported to be a differentially expressed lncRNA during influenza A virus infection and could influence viral replication and viral protein synthesis. 47 For the reasons mentioned earlier, it is possible that antisense lncRNAs and lincRNAs play pivotal roles in infection processes.
lncRNAs can trigger many biological effects in multiple processes. We used ENSG00000247095 to explore these potential biological functions. As shown in Figure 3, ENSG00000247095 is significantly differentially expressed in 293T cells after HFV infection. We retrieved specific information about this lncRNA from the Ensemble database and found that it is a lincRNA located in chromosome 11: 565,660–568,457. The transcript is called MIR210HG-003, has a length of 816 bp, and contains two exons. Some studies have shown that lncRNA can influence nearby genes to affect biological function. Based on this finding, we subsequently located nearby genes of ENSG00000247095, including phrf1 and rassf7. PHRF1 promotes TGF-β cytostatic signaling through TGIF ubiquitination at lysine 130. 48 In addition, RASSF7 negatively regulates pro-apoptotic c-Jun N-terminal kinase (JNK) signaling by inhibiting the activity of phosphorylated mitogen-activated protein kinase kinase (MKK7). 49 We hypothesize that lncRNA ENSG00000247095 function by regulating PHRF1 or RASSF7, but more experiments, including RNA pull-down and RNA immunoprecipitation, will be required to prove this hypothesis. This information could provide us with directions for future research, and other lncRNAs could imitate this example.
GO and KEGG pathway analyses suggest that lncRNAs may influence biological functions through these effects and mechanisms. Moreover, changes in a large number of lncRNAs are related to these effects in 293T cells after HFV infection. For example, GO enrichment analysis of lncRNA targets (Fig. 5B) showed that death-inducing signaling complex, T cell proliferation, and positive regulation of interleukin-8 (IL8) production are involved in lncRNA regulatory processes. lncRNAs might modulate the death-inducing signaling complex to influence host effects, leading to changes in viral replication or transcription. T cell proliferation is related to regulation of innate and adaptive immune responses. lncRNA may also play a role in positive regulation of IL8 production, which can modulate immune cells and inflammatory responses. GOTM (Fig. 5C–E) was further annotated with every GO number and the corresponding biological effects to help us find possible results more quickly. Then, lncRNA-targeted KEGG enrichment (Fig. 6B) showed several specific pathways to be considered mechanistically. According to the KEGG enrichment analysis, we can preliminarily ascertain pathways with either a significant difference or no significant difference. As with the pathway of cytokine–cytokine receptor interaction, we could design our project to further these findings.
Cis-acting and trans-acting regulatory networks may help us directly find key protein genes that lncRNAs might target. Data in RNA-seq show that lncRNAs could target different protein genes or different transcripts of the same protein gene in a cis-acting manner, but each protein gene is generally targeted by one lncRNA. In addition, we found that the possibility of correlation between lncRNAs and protein genes in a cis-acting manner is lower than in a trans-acting manner. In a cis-acting manner, for example, NR_028036 might target the fas gene, which encodes a protein belonging to a member of the TNF-receptor superfamily. Fas/FasL mainly mediates the death signaling pathway in antiviral immune response. In a trans-acting manner, we found that ENST00000354838 may target TLR7, which can participate in pathogen recognition and activation of innate immunity. We noticed that ENST00000354838 could target IL18, which can increase nature killer cell activity. These two manners illuminate the function of lncRNAs in antiviral immune responses and require validation in subsequent experiments.
In addition, we made some predictions of novel lncRNAs by using RNA-seq. We identified 58 novel lncRNAs that are located on most chromosomes. There are 21 upregulated and 37 downregulated novel lncRNAs. Among them, 5 upregulated lncRNAs are log2 ≥ 1, and 13 downregulated lncRNAs are log2 ≤ −1. Collectively, we anticipate that these data can further expand the number of known lncRNAs and that their functions will be discovered in future research.
In summary, we used next-generation sequencing to first show a complete map of lncRNA expression and to identify thousands of differentially expressed lncRNAs in 293T cells after HFV infection. In addition, we provided new insights into the mechanism of HFV infection and host response. More importantly, future research will be based on these data for better characterization of HFV infection.
Footnotes
Acknowledgments
This work was supported by the National Natural Sciences Foundation of China (nos. 81371790, 81641093, 81371422, 81571481, and 31170154), Major AIDS and Viral Hepatitis and Other Major Infectious Disease Prevention and Control project of China (2014ZX10001003), the Fundamental Research Funds for the Central Universities of China, and the Translational Medical Research Fund of Wuhan University School of Medicine.
Author Disclosure Statement
No competing financial interests exist.