
Abstract
Most HIV subtypes prevalent in China can be found in Shenzhen, including CRF07_BC, CRF01_AE, CRF08_BC, CRF55_01B, and subtype B. Multiple subtypes spreading in the same population always lead to the emergence of unique recombinant strains. Here, we report two unique recombinant forms (SZ44LS7251 and SZ95LS8027) of HIV-1 identified in a heterosexual population. Recombinant analyses were fulfilled based on the near full-length genomes. Both strains comprise subtypes B, C, and CRF01_AE. Phylogenetic analysis reveals that SZ44LS7251 is the second generation recombination originated from CRF55_01B andCRF07_BC, whereas SZ95LS8027 comprises CRF01_AE and CRF07_BC.The emergence of second generation recombination of HIV with complicated genomic structures supposed that high ratio of super infections or coinfections might happen in the Shenzhen area.
R
Shenzhen is one of the first cities that employed the Opening and Reform Policy in China. With much floating population, Shenzhen is favorable for spread of HIV. All of the dominant strains prevalent in China, including subtypes B, CRF01_AE, CRF07_BC, CRF08_BC, and CRF55_01B, can be found in Shenzhen, which is appropriate for the emergence of unique recombinant forms (URFs). 13
In this study, we characterized two HIV-1 genomic sequences in URF that comprised CRF01_AE, CRF07_BC, and CRF55_01B in Shenzhen. Both patients were infected through heterosexual contact. SZ44LS7251 was obtained from a 49-year-old patient, and SZ95LS8027 was obtained from a 38-year-old patient. Viral RNA was extracted from plasma samples using a high pure viral RNA kit (Roche, Basel, Switzerland). Viral near full-length genome was amplified in two halves using Platinum Taq DNA polymerase High Fidelity kit (Invitrogen, Carlsberg, CA) with more than 300 bp overlapping region using a nested reverse transcriptase PCR. PCR products were purified and sequenced by the Beijing Liuhe Huada gene technology company (BGI, Beijing, China) with a variety of internal specific primers (available on request).
The sequenced fragments were assembled with ContigExpress software (a component of Vector NTI version 11.5.1; Invitrogen). The final sequences of SZ44LS7251 and SZ95LS8027 were 8,808 bp length (from 790 to 9,604 according to the HXB2 calibrator) and 8,810 bp length (from 790 to 9,606 according to the HXB2 calibrator) separately. A Basic Local Alignment Search Tool (BLAST) search against the HIV-1 sequence database (
Two near full-length genomic sequences were aligned with HIV-1 strains of various reference subtypes (A–D, F–H, J, and K), and China prevalent recombinant forms (CRF01_AE, CRF07_BC, CRF5501_B, etc.) using Muscle software. The alignment was then edited manually in BioEdit software (version 7.0.0; T. Hall, North Carolina State University, Raleigh, NC). A maximum-likelihood (ML) tree was constructed by PhyML (
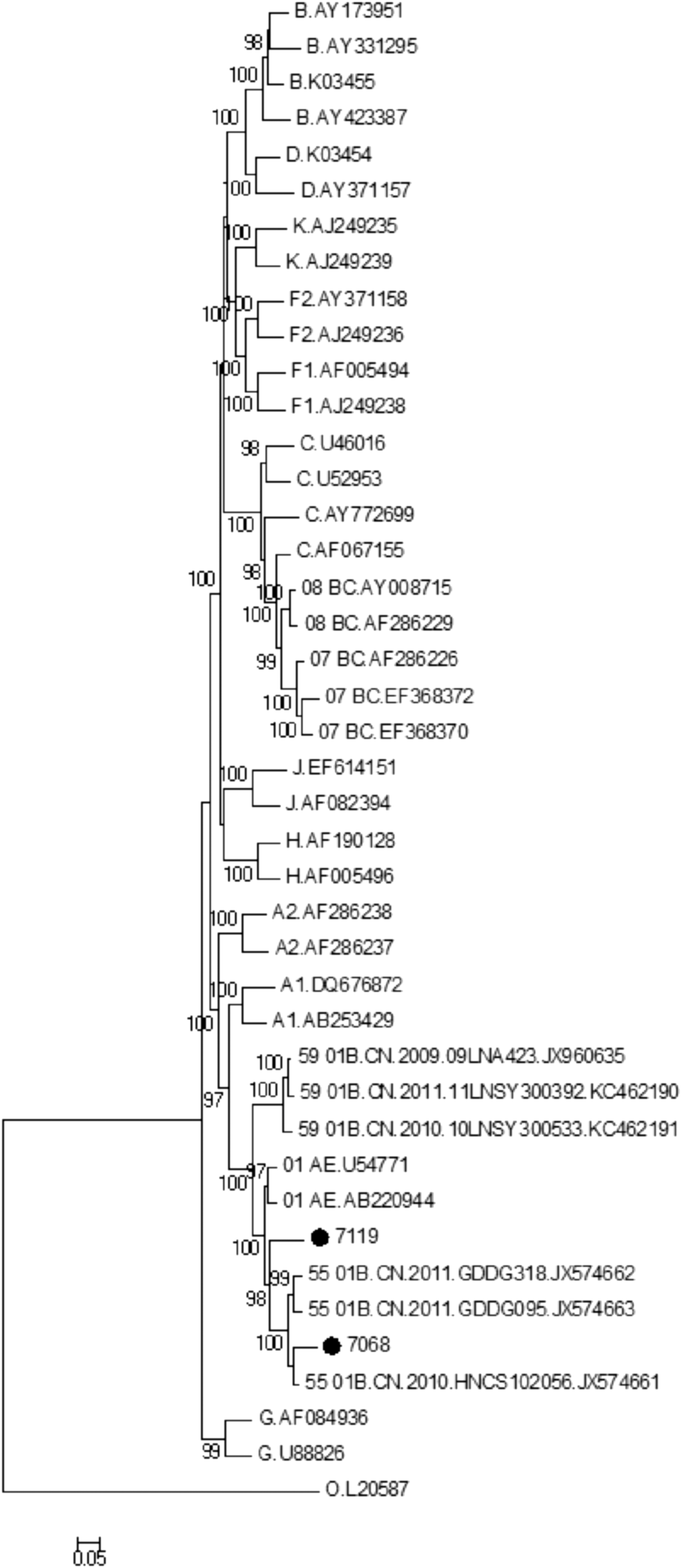
Phylogenetic analyses. NFLG nucleotide sequences of the patients SZ44LS7251 (7068, ●) and SZ95LS8027 (7119, ●) were aligned with HIV-1 strains of various reference subtypes and an ML tree was constructed by using the MEGA6 (Molecular Evolutionary Genetics Analysis Version 6.0; Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S) software package. All the reference strains were retrieved from the Los Alamos National Laboratory HIV Sequence Database (
Online software jpHMM-HIV was further performed to determine the break points and draw the genomic map. Eight break points were identified in SZ44LS7251, whereas five break points were identified in SZ95LS8027. Some similar break points were identified between these two sequences with CRF 07_BC (four break points) or CRF55_01B (three break points) separately (Fig. 2). The subtype of each segment was further verified using phylogenetic analysis with the reference sequence of subtypes A–D, F–H, J, and K and group O, which further proved the results of jpHMM analysis (data not shown).
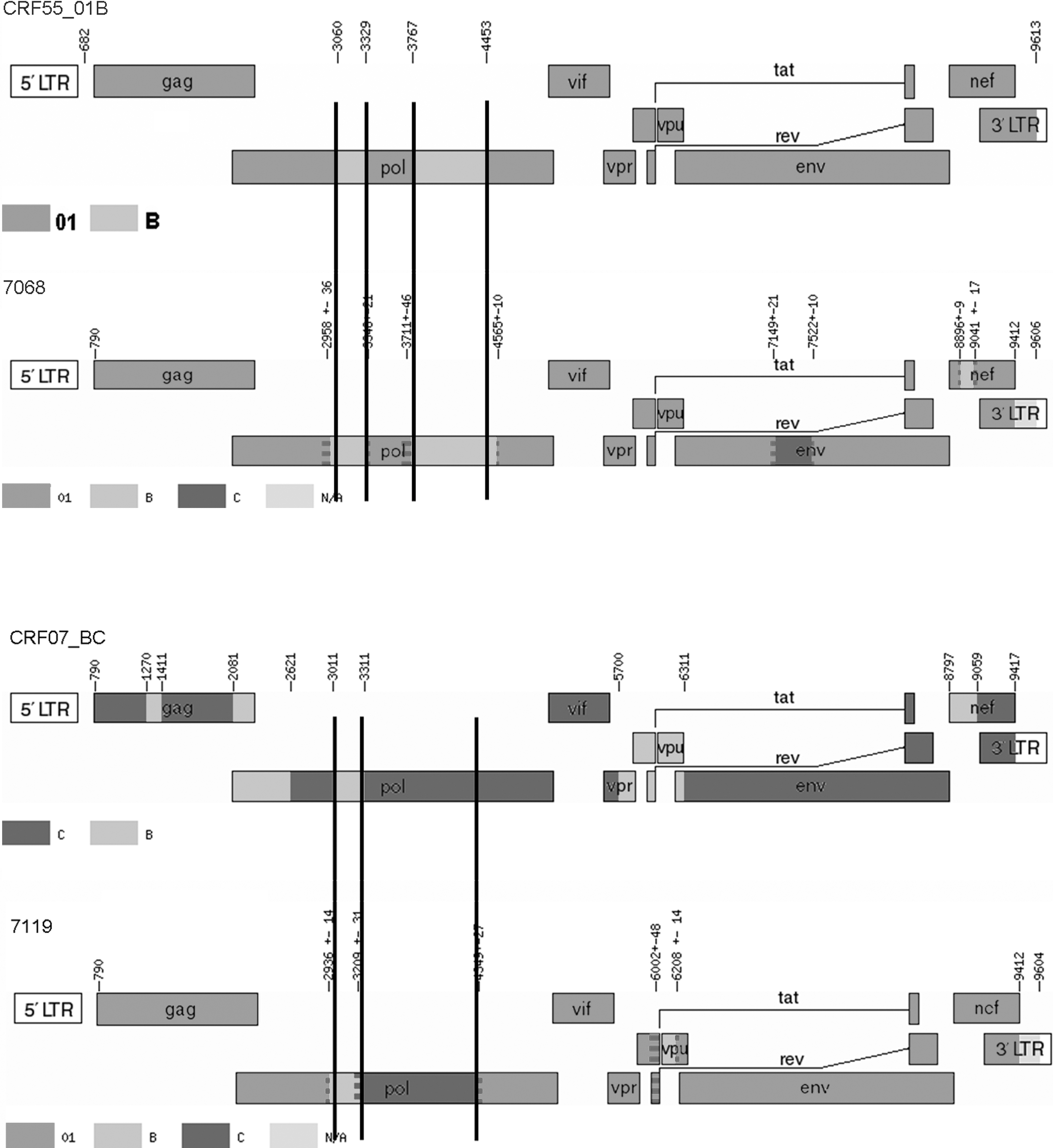
Genomic maps of SZ44LS7251 (7068) and SZ95LS8027 (7119). Numbers in the figure denote locations of recombination break points calibrated by HXB2. Genome maps of CRF 07_BC and CRF55_01B were downloaded from Los Alamos National Laboratory HIV Sequence Database (
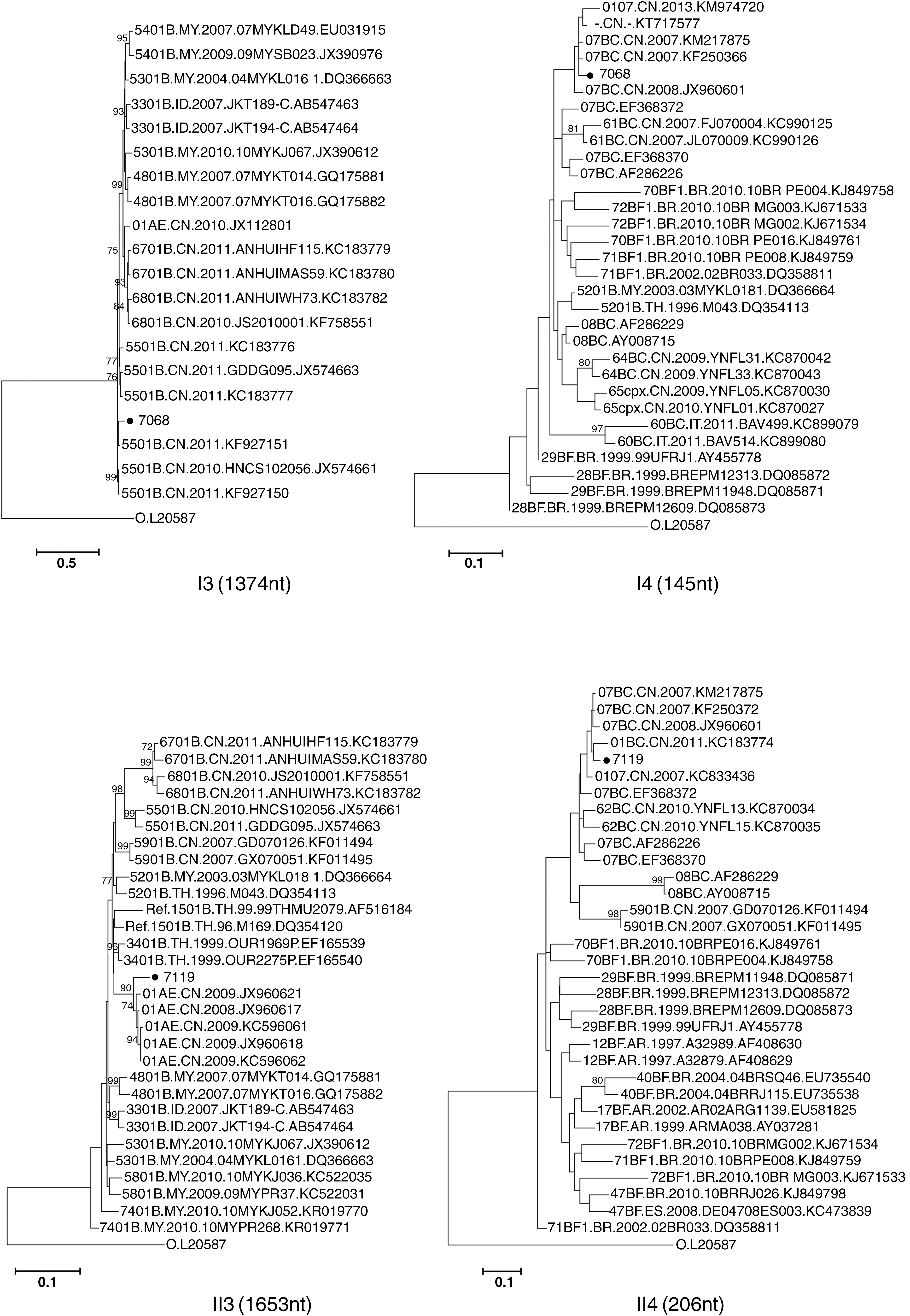
To explore the origin of each subtype region, phylogenetic analysis was further fulfilled based on each segment. BLAST search was used to obtain five high identity sequences from HIV Los Alamos sequence database. All of the CRFs that contained the same subtype segments were also included. The ML phylogenetic trees were constructed using the GTR + I + G or GTR + G substitution model that were selected with jModeltest software, the reliability of topologies was estimated by performing bootstrap analysis with 1,000 replicates (Fig. 3). For SZ44LS7251, all of segments clustered separately with CRF07_BC or CRF55_01B, supposing that it was their second generation recombination. For SZ95LS8027, all of segments clustered separately with CRF07_BC or CRF01_AE, supposing that it was their second generation recombination. CRF55_01B was estimated to be originated in Shenzhen a few years ago; the emergence of second generation recombination originated from it supposed that coinfections or super infections in some populations in Shenzhen should be monitored.

Phylogenetic analysis of different genomic segments. The span lengths of each segment were labeled separately. The possible phylogenetic origin of each segment was further verified by constructing ML trees. All of subtype reference sequences and most similar gene segments were obtained through BLAST search of Los Alamos database. Bootstrap analysis was performed with 1,000 replications and only bootstrap values ≥70% are shown.

In this article, we reported two novel recombinant strains identified in Shenzhen. Both of them are second generation recombination. In previous studies, most of the URFs were identified in injecting drug users or men who have sex with men, very few URFs were found in the heterosexual transmitted population. The identification of URFs in the heterosexual transmitted population in Shenzhen supposes that the HIV epidemic in the population is becoming serious. Heterosexual transmission has become the dominant route of spread of HIV in China. More than 60% new HIV infections have been caused by heterosexual transmission since 2011. Since the number of people in the general population who are involved in the heterosexual transmission is very large, the emergence of URFs among them urges the surveillance of HIV epidemic.
Footnotes
Acknowledgments
This work was supported by the National Key S&T Special Projects on Major Infectious Diseases (grant nos. 2012ZX10001-002 and 2012ZX10001-006) and the National Natural Science Foundation of China (no. 81273137).
Sequence Data
The nucleotide sequences of SZ44LS7251 and SZ95LS8027 have been submitted to GenBank with accession numbers KX378999 and KX379000.
Author Disclosure Statement
No competing financial interests exist.