
Abstract
In this study, the genetic diversity of HIV-1 in Tunisia was analyzed. For this, 193 samples were collected in different regions of Tunisia between 2012 and 2015. A protease and reverse transcriptase fragment were amplified and sequenced. Phylogenetic analyses were performed through maximum likelihood and recombination was analyzed by bootscanning. Six HIV-1 subtypes (B, A1, G, D, C, and F2), 5 circulating recombinant forms (CRF02_AG, CRF25_cpx, CRF43_02G, CRF06_cpx, and CRF19_cpx), and 11 unique recombinant forms were identified. Subtype B (46.4%) and CRF02_AG (39.4%) were the predominant genetic forms. A group of 44 CRF02_AG sequences formed a distinct Tunisian cluster, which also included four viruses from western Europe. Nine viruses were closely related to isolates collected in other African or in European countries. In conclusion, a high HIV-1 genetic diversity is observed in Tunisia and the local spread of CRF02_AG is first documented in this country.
S
In this study, we investigate HIV-1 infection in Tunisia in samples collected during the past 3 years, with the objective of updating the information on HIV-1 genetic diversity in the country. For this purpose, we obtained 193 samples from HIV-1-infected individuals collected through our routine and research activities in Tunisia between 2012 and 2015. In Tunisia, since the notifications of the first cases of HIV/AIDS until October 31, 2015, a total of 2,159 cases of HIV-1 infection were registered, of which 612 correspond to persons who have died. The HIV prevalence in Tunisia is 14.4/100,000 inhabitants, which has remained relatively stable over recent years, placing Tunisia among the low prevalence countries. The main mode of transmission is through heterosexual contact (45.34%), followed by injecting drug use (21.44%), mother to child transmission (4.58%), and homosexual contact (5.32%), and for 18.2% the transmission route is unknown [Primary healthcare directory (DSSB), Ministry of Health, Tunisia].
Whole blood specimens from four infectious disease centers (located in Tunis, Sfax, Sousse, and Monastir) in Tunisia were sent to the microbiology laboratory of Charles Nicolle hospital, in Tunis, where they were processed. A pol fragment comprising protease and the 5′ segment of reverse transcriptase was amplified from RNA extracted from plasma and sequenced using a published protocol
11
or the protocol of the National Agency for AIDS Research (
The obtained sequences, ∼1.3 kb long, were aligned with reference sequences of subtypes and CRFs using MAFFT v.7.
12
Phylogenetic trees were constructed through maximum likelihood with RAxML v.7.2.7
13
using the general time reversible with gamma-distributed among-site rate heterogeneity (GTR+Γ) substitution model. To examine possible intersubtype mosaicism, sequences were analyzed using the Recombination Identification Program (RIP) available at the HIV Sequence Database site (
All sequences obtained in this study were deposited in GenBank, with accession numbers KM006229-KM006263 and KX183686-KX183846.
The analysis of the protease-reverse transcriptase region from 193 HIV-1-infected individuals from Tunisia revealed that subtype B is the most prevalent genetic form (n = 90; 46.4%), followed by CRF02_AG (n = 76; 39.4%). Nine other HIV-1 circulating genetic forms were identified: subtypes A1 (n = 3; 1.6%), C (n = 3; 1.6%), D (n = 2; 1%), G (n = 2; 1%), and F2 (n = 1; 0.5%) and CRFs 06_cpx (n = 2; 1%), 25_cpx (n = 1; 0.5%), 43_02G (n = 1; 0.5%), and 19_cpx (n = 1; 0.5%) (Fig. 1). Eleven URFs were also identified through bootscanning: three CRF02_AG/B, with different break point positions, and one each of B/D, A1/G, B/G, A1/U, A1/F1/G, CRF13_cpx/U, A1/B/G, and A1/B/CRF02_AG (Fig. 2). Among CRF02_AG viruses, 44 formed a cluster supported by a high bootstrap value (Fig. 1).
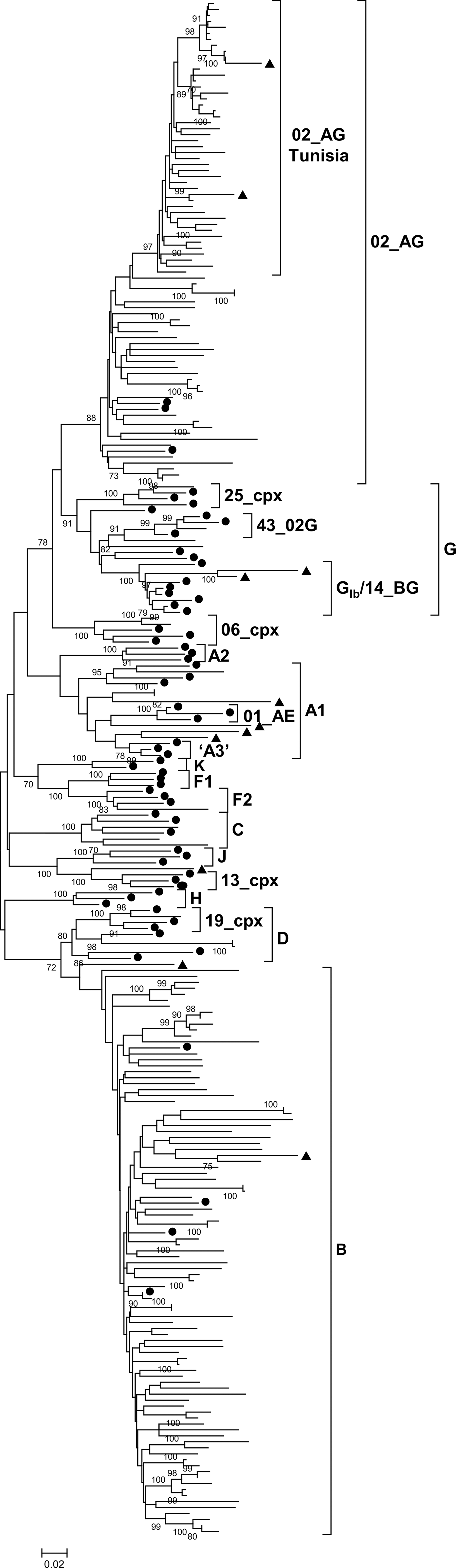
Maximum likelihood phylogenetic tree of 193 HIV-1 PR-RT sequences from Tunisia. Samples from Tunisia are unlabeled. Circles denote subtype and CRF references. Triangles denote sequences that upon subsequent analysis were shown to be URFs (Fig. 2). Only bootstrap values of 70% or greater are shown. CRF, circulating recombinant form; URFs, unique recombinant forms; PR-RT, protease-reverse transcriptase.
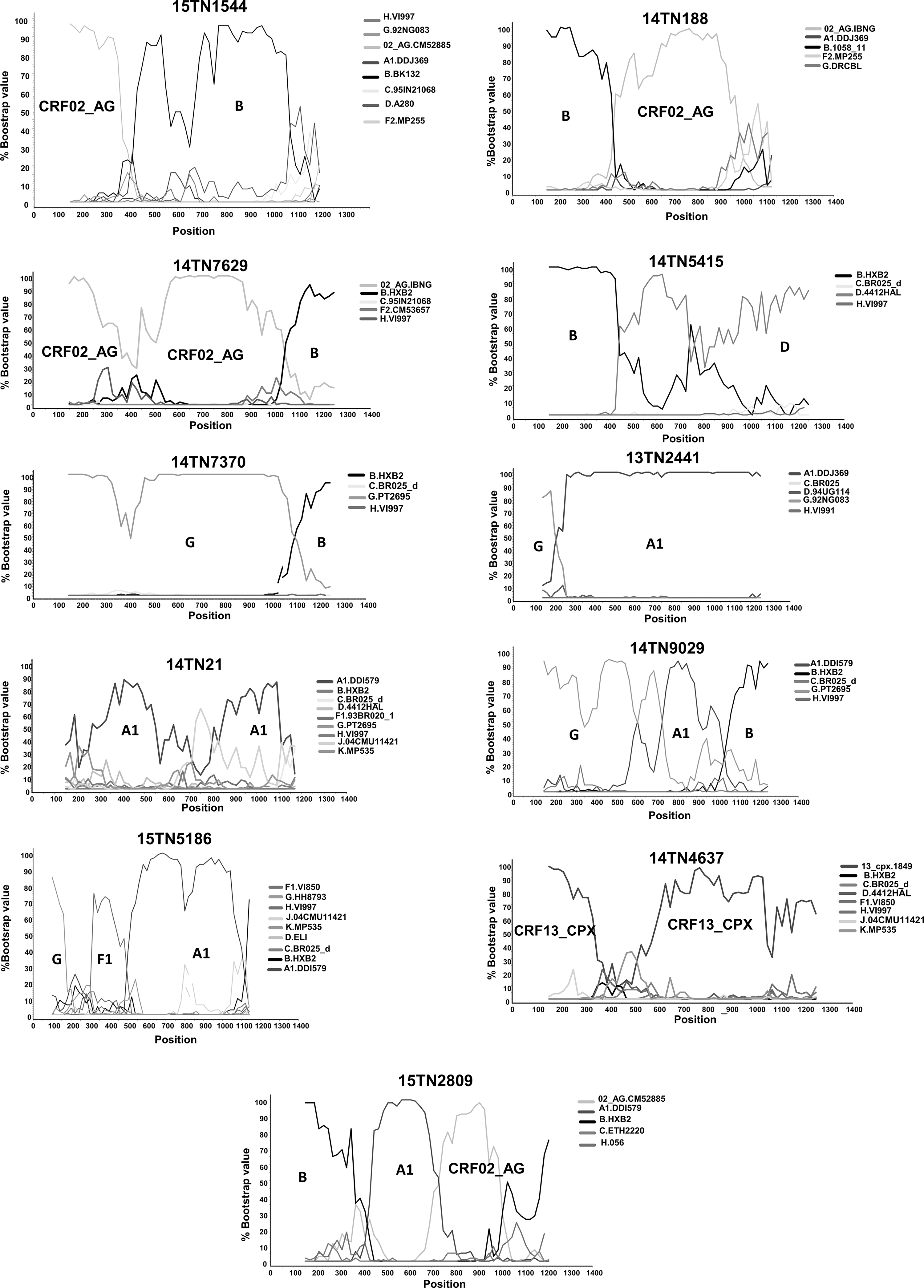
Bootscan plots of PR-RT sequences of 11 HIV-1 URFs identified in Tunisia.
Through similarity searches in databases and subsequent phylogenetic analyses, we found that nine viruses from Tunisia were closely related to viruses from Algeria, Chad, Czech Republic, Nigeria, United Kingdom, and Romania (Fig. 3). We also found that four viruses collected in western European countries (Sweden, Denmark, and Spain) branched within the Tunisian CRF02_AG cluster (Fig. 3). Notably, CRF02_AG viruses from other North African countries failed to branch with the Tunisian cluster (data not shown).
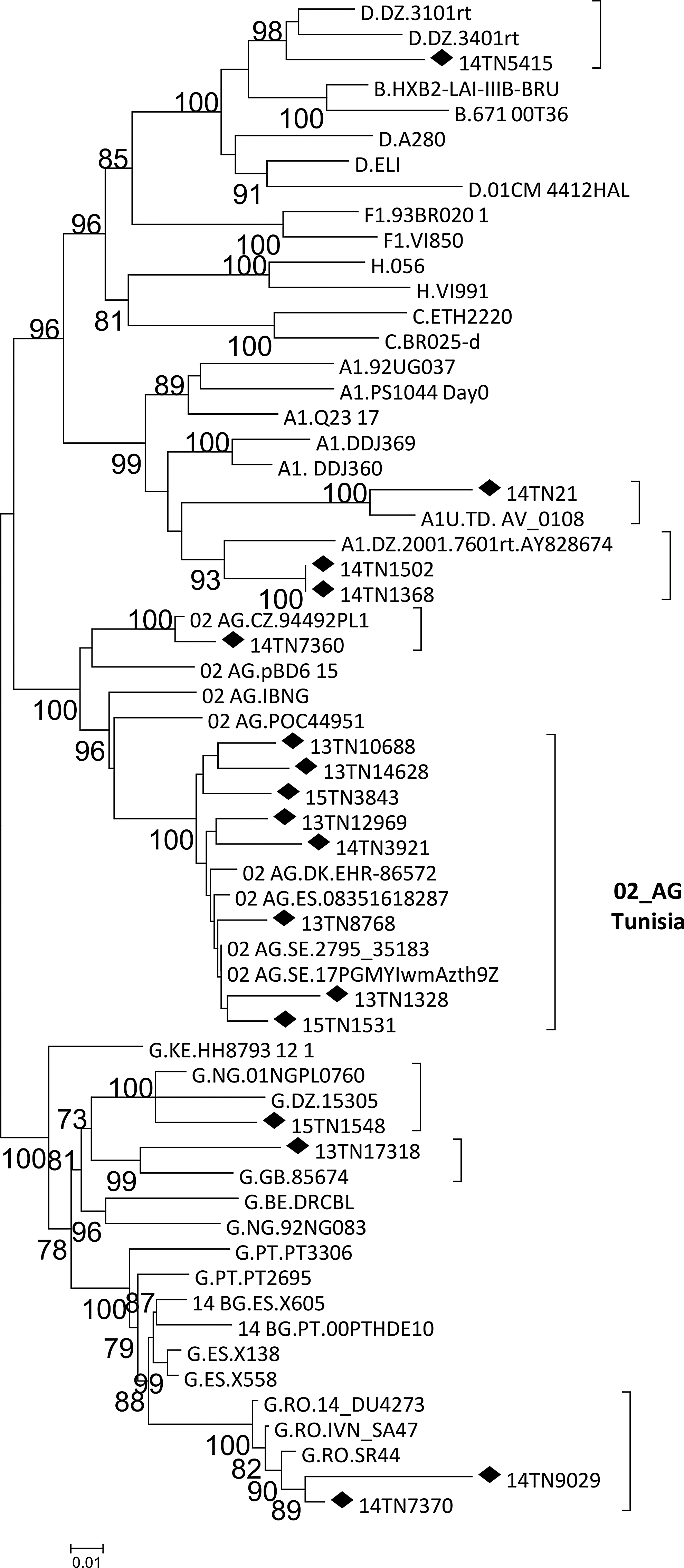
Phylogenetic tree showing the relationship between HIV-1 isolates from Tunisia and database viruses. Sequences from Tunisia are labeled with diamonds. Countries of collection of database sequences are indicated with the two-letter ISO country code. Only bootstrap values 70% or greater are shown.
The distribution of HIV-1 genetic forms is less well known in North Africa than in sub-Saharan Africa because of the relative scarcity of data. 8,15 –17 With regard to Tunisia, very little was known about the HIV-1 genetic diversity due to the small sample size of the only published study 10 and the lack of recent data. In the Middle East and North African region, a great genetic diversity of HIV-1 has been observed with heterogeneous subtype and CRF distribution. 16 In a previously published study, subtype B was largely predominant in Tunisia (95%) with one sporadic CRF02_AG case (4.7%), 10 similarly to what has been reported in Morocco. 17 In a recent study in Algeria, CRF06_cpx was found to be predominant (72%). 9 In this study, we found a notable increase in the prevalence of CRF02_AG in Tunisia, which is greater than in other North African countries. However, subtype B is still the predominant clade, as in Morocco. 6
Interestingly, 44 (57.8%) CRF02_AG Tunisian viruses formed a separate cluster (Fig. 3), with no sequences from other North African countries included in it. Therefore, this cluster is specific of Tunisia. In addition, we also detected sporadic infections with five other subtypes and four other CRFs in Tunisia. The explanation for the current high diversity of HIV-1 clades and the recent changes observed is probably multifactorial, including an increase in migration due to the economic and social situation in Tunisia and neighboring countries. Interestingly, we identified several URFs in a minor proportion (5.6%). Some of these derive from clades not circulating in Tunisia and are probably imported from other countries. However, three URFs derive from recombination between the two main clades circulating in Tunisia (B and CRF02_AG), suggesting a Tunisian origin, and since break points were different, they indicate the existence of dual infections with different HIV-1 clades and active recombination in Tunisia.
The great diversity of HIV-1 genetic forms detected in this study probably reflects the impact of migration from West Africa to Tunisia as a transit country for subsequent migration to Europe, which increases the genetic complexity of the HIV-1 epidemic in Maghreb and in Europe. These changes reflect a growing trend, also observed in other studies, showing an increase in the proportion of CRFs/URFs in North Africa in the past 15 years. 6,15 Therefore, classifying or determining the origins of a presumed CRF/URF is particularly challenging in Tunisia, where subtypes and intersubtype recombinant forms are cocirculating.
Phylogenetic clustering indicates that some viruses detected in Tunisia are closely related to viruses from Algeria, Chad, Czech Republic, Nigeria, United Kingdom, and Romania. Furthermore, viruses from Sweden, Denmark, and Spain branched within the Tunisian CRF02_AG cluster. Whether this reflects migration of HIV-1-infected Tunisian individuals to Europe or infection in Tunisia of Europeans while visiting the country is not known, given the lack of epidemiological information for the database viruses.
In summary, we found a great diversity of HIV-1 genetic forms in recently collected samples from Tunisia, with predominance of subtype B and CRF02_AG, with 57.8% CRF02_AG viruses forming a Tunisian cluster. Recent socioeconomic developments promoting migration from sub-Saharan Africa to western Europe, using Tunisia and other North African countries as a transit area, could explain the increase in CRF02_AG and various other clades observed in this study. However, it is uncertain whether biological properties might have played a role in the spread of CRF02_AG in Tunisia. The challenges posed by the changes in the genetic composition of the HIV-1 epidemic in Tunisia and other North African countries, which is becoming increasingly complex, highlight the need to promote surveillance studies on HIV genetic diversity in this region.
Footnotes
Acknowledgments
We thank the personnel at Infectious Diseases Centres in Tunis, Monastir, Sousse, and Sfax for their support of this study. Also we thank all the laboratory staff for their excellent technical assistance in HIV Biology and Variability Unit, Centro Nacional de Microbiología, Instituto de Salud Carlos III, Majadahonda, Madrid, Spain, and in Unit Virology, Microbiology Laboratory, Charles Nicolle University Hospital, Tunis, Tunisia. This work was funded by the Global Fund to Fight AIDS, Tuberculosis, and Malaria.
Author Disclosure Statement
No competing financial interests exist.