
Abstract
HIV-2 exhibits a natural history of infection distinct from HIV-1. Primarily found in West Africa and in only 10%–20% of HIV infections in this region, patients with HIV-2 typically exhibit a slower progression to AIDS, lower viral loads, and decreased rates of transmission. Here, we used next-generation sequencing to determine the sequence and phylogenetic classification of nine HIV-2 genomes. We identified a patient with a series of mutations in an invariant cytotoxic lymphocyte (CTL)-restricted gag epitope required for retroviral structure and replication and implicated in long-term nonprogression to AIDS. The presence of wild-type sequence argues these mutations are involved in immune escape, whereas its reversion to a sequence seen only in the sooty mangabey reservoir suggests an alternate means of controlling infection. Surveillance and molecular characterization of circulating strains are essential for continued development of monitoring tools and may provide greater insight into the reduced pathogenicity of HIV-2.
Introduction
H
Increased molecular characterization of strains would greatly benefit research into the distinct pathogenesis of HIV-2. Currently, only 30 full-length HIV-2 genomes are present in databases compared with 5,927 for HIV-1. 6 There is also no regulatory-approved viral load test due, in part, to the lack of reference materials to assess assay performance. 7 Here, we report nine full-length HIV-2 genomes obtained by unbiased next-generation sequencing (NGS) and identify a virus isolate with a series of mutations in an immunodominant epitope linked to long-term nonprogression.
Materials and Methods
Virus isolates
HIV-2 strains were collected by the French National Reference Center for HIV from West African patients living in France and propagated in cell culture as previously described. 8 Preliminary strain classification was based on reverse transcriptase-polymerase chain reaction amplification of RNA extracted from supernatant followed by population sequencing by Sanger and phylogenetic analysis of subgenome regions of gag, pol, and env as previously described. 9
RNA extraction
Culture supernatants were diluted at 1:100 v/v in NHP, and automated RNA extractions were performed on an m2000sp instrument using the RNA Sample Preparation System (Abbott Molecular, Des Plaines, IL) according to the manufacturer's instructions; sample volumes were 200 μl and 500 or 600 μl with elution volumes of 100 and 70 μl, respectively. Resulting RNA inputs for each cDNA reaction are listed in Table 1.
HIV-2 phylogenetic classification and sequencing statistics are listed for each strain.
Viral loads
Cell culture supernatant was diluted into HIV-uninfected normal human plasma (NHP), and viral load was measured on the m2000 using a research assay as previously described, except that a pumpkin-derived RNA was used as an internal control, and RNA extracted from electron microscopy-quantified virus particles of HIV-2 NIH-Z strain was used as the HIV-2 calibrator. 10
NGS library synthesis
NGS libraries of HIV-2 virus isolates were prepared with the Ovation Single Cell RNA-Seq System (NuGen). In brief, reverse transcription is initiated with a combination of oligo(dT) and random primers targeting non-rRNA sequences. First-strand cDNA synthesis randomly incorporates a degradable nucleotide analog. A subsequent processing step degrades this analog and the template RNA to yield single-stranded cDNA fragmented to an average size of 230 nt. Random octamers fused to the forward adaptor direct second-strand synthesis. After end repair, reverse adaptors containing unique barcodes are ligated to the free end to create double-stranded cDNA. Two successive rounds of purification on RNAClean XP magnetic beads and PCR amplification steps (of 14 and 13 cycles) are then performed. Library concentrations were measured on a Bioanalyzer 2200 TapeStation using a D1K ScreenTape (Agilent Technologies, Santa Clara, CA) and adjusted to 1 nM before multiplexing. Libraries were combined in equal volumes, denatured with 0.1 N final NaOH for 5 min, and diluted to 20 pM with HT1 buffer. The multiplex library was diluted once more with HT1 to 12 pM, and 1% PhiX loading control was added. The multiplex library was denatured at 96°C for 2 min, snap chilled on ice, and then run on a MiSeq instrument using a 300 cycle MiSeq Reagent Kit v2 (Illumina). Paired-end reads were sequenced, but only one barcode was read.
NGS analysis
Sample barcodes were parsed on the MiSeq instrument, and reads were filtered for Q-scores above 30. CLC Genomics Workbench 8.0 software (CLC bio/Qiagen, Aarhus, Denmark) was used for analysis, and reads were initially aligned to the 2A-BEN reference strain at previously described settings, followed by iterative rounds of refinement by mapping to the consensus genome. 8 The raw data were realigned to the final consensus sequence to generate the NGS statistics. Open reading frames were verified and annotated using SeqBuilder (DNASTAR Lasergene v11.2). Full-genome consensus sequences have been deposited into GenBank under the following accession numbers: KU168287 (LA36), KY025538 (LA37), KY025539 (LA38), KY025540 (LA39), KY025541 (LA40), KY025542 (LA41), KY025543 (LA42), KY025544 (LA43), KY025545 (LA44).
Phylogenetic analysis
Final genomic sequences were merged into an alignment of 30 full-length HIV-2 sequences present in the Los Alamos HIV database. 6 Analysis in PHYLIP software and generation of neighbor-joining trees were performed as described. 8
Results
Full-genome sequencing of HIV-2 by NGS
Although HIV-2 has the same viral life cycle as HIV-1 and also causes AIDS, titers in infected individuals are generally much lower and therefore create a challenge for molecular characterization directly from patient plasma. To facilitate sequencing, HIV-2 strains obtained from West African patients residing in France were propagated on activated human PBMC in cell culture. After viral load measurement of isolates on the m2000rt, ∼150–16,000 copies of virus were used as input for randomly primed cDNA synthesis by the Ovation Single Cell RNA-seq System (Table 1). Nine individual libraries were barcoded and sequenced together on a single MiSeq run. Full-genome (100%) coverage was obtained for each strain, with the number of HIV-2 mapped reads ranging from 23,895 to 889,037 and the sequencing depth ranging from 332 × to 12,314 × (Table 1). Low standard deviations relative to the mean were indicative of the uniformity in coverage obtained by this method.
Consensus genomes were compared to full-length sequences obtained by an alternate NGS method or to subgenomic fragments (gag/pol) obtained by population sequencing with the Sanger method to verify the results (Ref., 8 data not shown). As an example, the LA36 genome obtained in this study differed by five nucleotides (99.99% identity) from the HIV-SMART-generated sequence (10,007 nt); Sanger sequences for gag (540 nt) and pol (1,019 nt) were 100% and 99.6% identical, respectively. Genomes were merged into an alignment of full-length HIV-2 sequences available in the Los Alamos database, and a neighbor-joining phylogenetic tree was constructed (Ref., 6 Fig. 1). Seven strains (LA36–LA42) branched with HIV-2 group A, while two (LA43, LA44) branched with HIV-2 group B. The addition of these nine HIV-2 sequences increases the database of full-length entries by 30%.
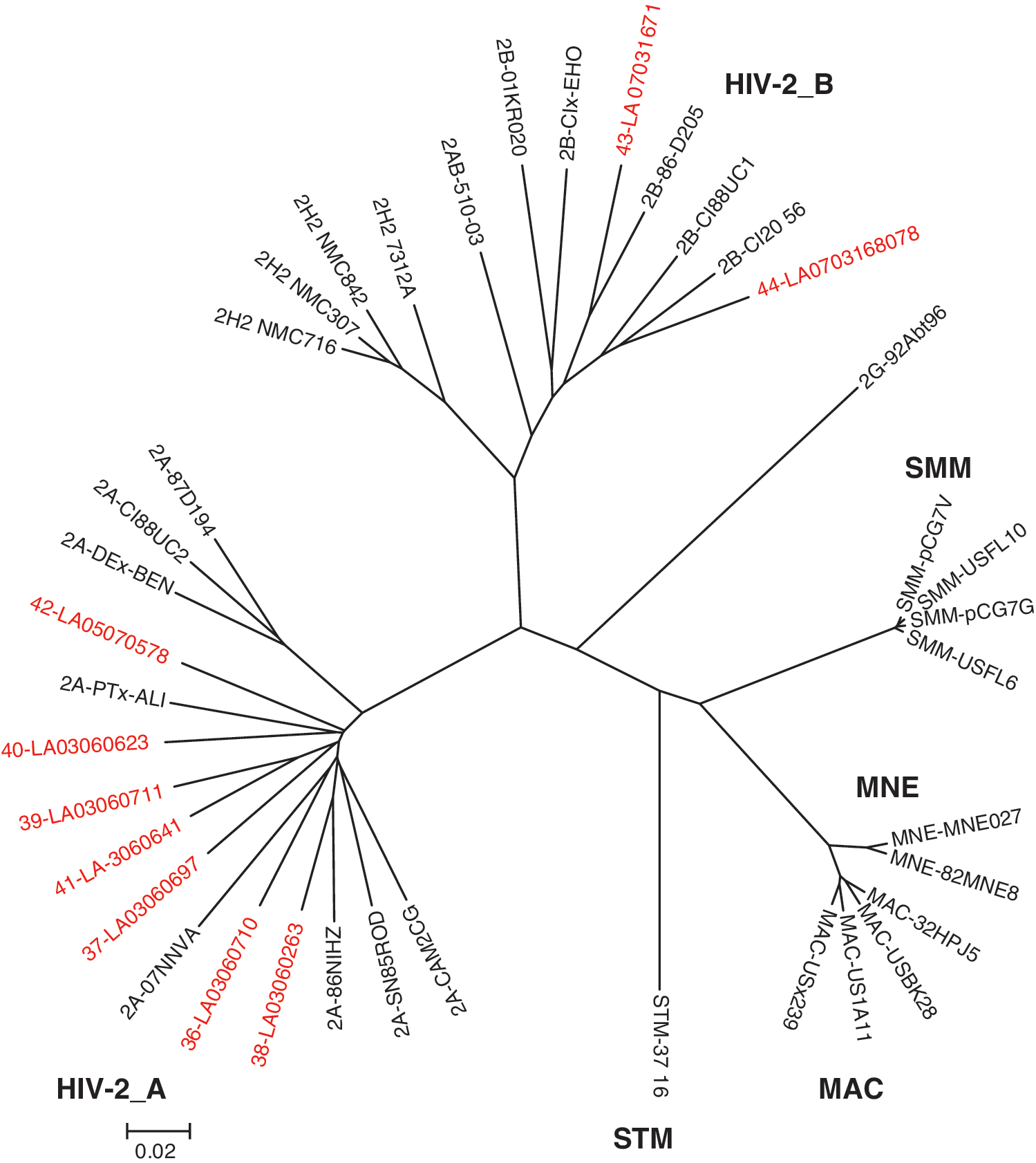
A neighbor-joining phylogenetic tree of available full-length HIV-2 and SIV sequences. New virus isolates are shown in red.
Mutations identified in an HLA-B14-restricted epitope of gag implicated in HIV-2 long-term nonprogression
Genomic sequences, including and surrounding an immunodominant CTL-restricted epitope implicated in HIV-2 long-term nonprogression to AIDS, were analyzed in the nine virus isolates. 11,12 Situated in the highly conserved major homology region of gag-p26 required for retroviral structure and replication, the 165DRFYKSLRA173 nonamer (DA9) in all except one of the nine new strains sequenced in this study matches the HIV-2 consensus (Fig. 2A). LA43 has a Ser->Gly substitution to yield the following peptide: 165DRFYKGLRA173. The corresponding amino acid at the sixth position in the majority of HIV-1 strains is Thr, the sole difference in this epitope that distinguishes it from HIV-2 (Fig. 2A). Four additional, novel substitutions (LEQS) relative to the HIV-2 consensus were also present in LA43 immediately 3′ of this epitope. Among these, a Pro required for optimal peptide cleavage by the immunoproteasome was substituted with Gln. 11 Notably, a Gln is typically found in HIV-1 at this position (Fig. 2A). NGS coverage depth in this region was 1,292-fold, and the exact same sequence was obtained by the Sanger method (Fig. 2B).
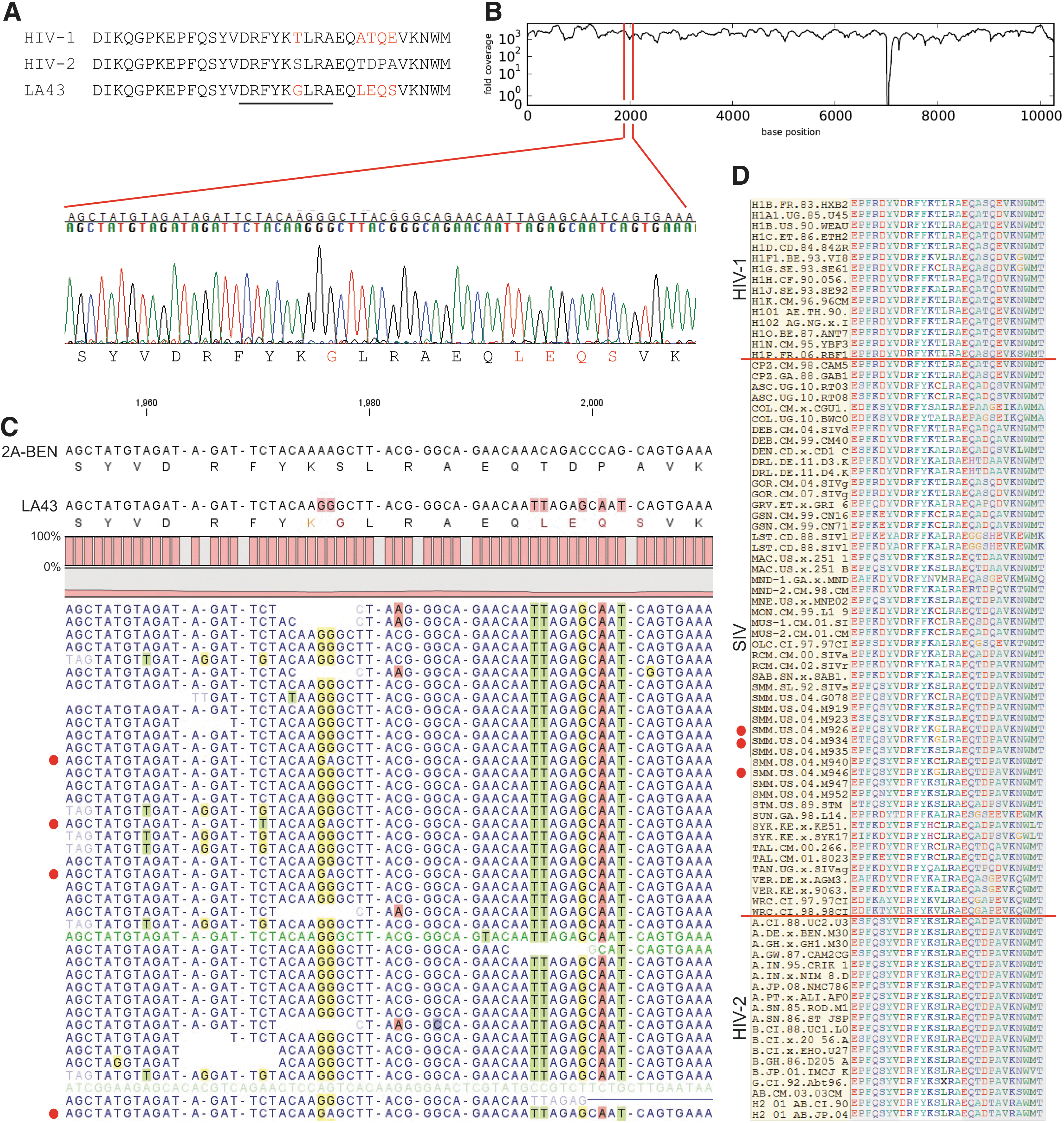
Inspection of NGS reads mapped to the epitope revealed heterogeneity within the Gly codon (GGC) at the sixth position. A minority (1%) of reads contained AGC at this position, translated as Ser (Fig. 2C). Considering remnants of this “wild-type” sequence (AGC) are still present suggests the Ser->Gly mutation in LA43 may have been selected to escape CTL pressure. Mapped reads of the downstream LEQS motif do not show any evidence that the TDPA sequence found in the majority of HIV-2 strains is present in this strain (Fig. 2C).
These motifs have not been described to date. Of the 239 HIV-2 gag entries in the Los Alamos National Lab database that cover this region, the DA9 nonamer is invariant for all positions, with the exception of four strains that replace Ser at position 6 with Gly (1; Ghana), Asn (1; Guinea-Bissau), or Cys (2; both Guinea-Bissau). 13 –15 The origin of LA43 is unknown, however, it branches most closely with a different H2B strain (2B-86-D205; Fig. 1) from Ghana. To understand if there was any precedent for the changes seen in LA43, an alignment of this epitope, including HIV-1 and SIV sequences from nonhuman primates (NHP), was examined. Interestingly, while multiple NHP species tolerate Ala, Cys, and Val at this position, as do some HIV-1 subtypes, Gly was only observed in sooty mangabey SIV sequences (Fig. 2D). 16 There is somewhat more variability present in the flanking region of the nonamer in HIV-2, but the substitution of the TDPA motif with LEQS was not observed in HIV-2, SIV, or HIV-1 sequences.
Discussion
A metagenomic approach provided full-length sequences and phylogenetic classification for nine HIV-2 virus isolates. With a limited number of complete genomes currently available, the extent of HIV-2 sequence diversity is uncertain and hampers optimization of diagnostic tests. Continued surveillance, capitalizing on the depth of sequence coverage afforded by NGS to uncover minor variants and dual infections, will provide investigators a measure of sensitivity not previously available.
The heterogeneous sequences mapping to the gag CTL-restricted epitope in LA43 was found by this means and raises several intriguing questions. Unfortunately, this specimen dates back to the late 1990's and the patient was lost to follow-up. No serial bleeds of the original plasma remain to determine if viral loads increased after acquisition of this mutation, nor are there cell pellets for HLA typing to determine if the virus did indeed evade the MHC class I restriction. Given that the majority (99%) of LA43 reads has the Ser->Gly mutation and that this sequence is found in one other individual from Ghana and in SIV from its zoonotic reservoir, sooty mangabey monkeys, viral fitness would not appear to be diminished. 14,16 While substitution with a G at position 6 was not directly tested in vitro in Wagner et al., similarly conservative Thr->Ala and Thr->Ser changes still yielded replication-competent viruses, and Ala, Val, Cys, and Iso amino acids are frequently observed in nature at this position, both in HIV-1 and SIV strains (Fig. 2D). 6,12 These changes in the HIV-1 nonamer were coincident with reduced CTL recognition and an ability to sensitize cells to lysis, potentially related to antigen presentation, and consistent with an inability to suppress HIV-1 replication. 12 Amino acid changes in regions flanking an epitope can either reduce or increase a CTL response, however, it would be interesting to explore if the peptide is also processed less efficiently as a result of the downstream sequence changes (e.g., Pro->Gln) and whether this, too, leads to a reduction in CTL responses (e.g., proliferation, IFN-γ production) and higher viral loads in a matched host. 11,17 Alternatively, as this nonamer sequence is found only in sooty mangabey monkeys, LA43 may represent a virus better adapted to its original host and in turn less pathogenic to humans. Indeed, the Ghana patient (GK030) with the Ser->Gly substitution is noted as being clinically asymptomatic. 14
With the nearly 37 million people infected with HIV-1 and 1.2 million dying annually from AIDS-related illnesses, more research is needed on HIV-2 to understand how a majority of people infected with this virus, sharing 30%–60% homology to HIV-1, come to acquire protective immunity, while those with HIV-1 fail to control the infection. Deploying NGS in the quest for answers will undoubtedly provide mechanistic insight into the biology behind this disparity.
Footnotes
Acknowledgment
We thank Dr. Sarah Rowland-Jones at the University of Oxford for helpful discussions.
Author Disclosure Statement
J.Y., C.A.B., G.A.C., and M.G.B. are all employees and shareholders of Abbott Laboratories. The other authors report no conflicts of interest.