
Abstract
An increasing amount of evidence suggests that HIV replication persists in gut-associated lymphoid tissues (GALT), despite treatment with combination antiretroviral therapy (cART). Residual replication in this compartment may propagate infection at other sites in the body and contribute to sustained immune dysregulation and delayed immune recovery. Therefore, it is important to focus efforts on eliminating residual replication at this site. There are several challenges to accomplishing this goal, including low antiretroviral (ARV) exposure at specific tissue locations within GALT, which might be overcome by using the tools of clinical pharmacology. Here, we summarize the evidence for GALT as a site of residual HIV replication, highlight the consequences of persistent infection in tissues, identify current pharmacologic knowledge of drug exposure in GALT, define the challenges that hinder eradication from this site, and propose several avenues for pharmacologic intervention.
Introduction
T
Utilizing decay rates of HIV RNA in plasma and DNA in peripheral blood mononuclear cells (PBMCs), Perelson et al. developed a mathematical model that estimated the overall half-life of viral decay to be ∼44 months. 6 Given this extremely long rate of viral decay, it has been estimated that it would take 73 years of cART to fully eradicate HIV from the body. 7 Therefore, HIV persistence despite cART is a hallmark of HIV infection and represents a significant barrier to cure.
It is well established that HIV latency is a primary driver of persistence in humans. The latent reservoir, comprising long-lived memory and potentially follicular helper T cells, is established early in infection and is unaffected by current antiretroviral (ARV) therapies. 8,9 However, it has also been hypothesized that ongoing viral replication from productively infected CD4+ T cells in certain tissue reservoirs may contribute to persistence.
Although this hypothesis remains controversial, mounting evidence suggests that active replication may be occurring in sites such as the central nervous system, genital tract, and lymphoid tissue. 10 –13 Gut-associated lymphoid tissues (GALT) is perhaps the most likely source of ongoing HIV replication given its high concentration of HIV target cells and its role as a site of initial HIV exposure and early infection. Given the extensive distribution of GALT and its important role in immune function, ongoing replication in this compartment may have clinical consequences that are not observed from other tissue reservoirs.
As the evidence for ongoing replication in tissue reservoirs continues to grow, so does the need for interventions that are aimed at eradicating HIV from these sites. Clinical pharmacology can play a large role in understanding how these reservoirs persist in the face of cART, and in the development of targeted interventions for HIV eradication. The tools of pharmacology can also help to clarify the mechanisms of persistence in GALT (e.g., active replication, latency, or both). The pharmacologic mechanisms influencing HIV tissue reservoirs have been previously reviewed, 14,15 but none have focused on the unique challenges faced in eradicating HIV replication in GALT. The purpose of this article is to summarize the evidence for ongoing HIV replication in GALT, address the challenges associated with current eradication strategies, and propose opportunities for pharmacologic interventions.
HIV Persistence in GALT
GALT is the largest component of the lymphoid system, comprising the tonsils, Peyer's patches, lymphoid aggregates in the stomach and small intestine, and lymphoid cells in the lamina propria. GALT contains the highest concentration of CD4+ T cells, making it an ideal target for HIV infection. 16,17 GALT is also one of the first tissues to become infected after exposure, with T cell decreases observed as soon as 4 days after infection. 17 –19 Given this early establishment of infection and the large number of target cells, this compartment may be a natural candidate for HIV persistence.
Clinical observations support this hypothesis, where HIV RNA shedding from the rectum of HIV-positive, sexually-transmitted infection-uninfected men was reduced but not eliminated with cART. 20,21 In addition, gut immune activation after initial infection has also been shown to persist despite long-term treatment, suggesting persistent exposure to viral antigens. 22 Further, the amount of infectious virus in active CD4+ T cells (many of which are located in the GALT) was found to be 1.6-fold higher than in resting CD4+ T cells, suggesting that latently infected cells do not account for all of the residual virus. 23
Given the lymphoid nature of GALT and the corresponding clinical evidence of persistent replication at this site, several groups have looked for molecular evidence of HIV persistence and compartmentalization in the gut. HIV RNA and DNA have been shown to concentrate in the gut compared with PBMCs, and this distribution changes along the length of the gastrointestinal tract (e.g., unspliced RNA in the ileum and rectum were increased 10.2- and 2.4-fold over blood, respectively). 24 In addition, the ratio of RNA/DNA (as high as 4.6) throughout the gut suggests low-level replication. 24 Additional work found that HIV DNA concentrations were on average fivefold higher in the gut versus PBMCs in patients on suppressive cART, even after correcting for T cell population differences between these compartments. 25
In a study in which DNA was isolated from the rectum, PBMCs, and plasma of patients not receiving cART, greater diversity was observed among gut-derived HIV variants versus those derived from PBMCs, and these variants were interspersed among all three compartments, suggesting movement between sites. 26 Further, no evidence of compartmentalization was observed in a study using DNA isolated from the ileum, colon, and PBMCs of HIV-positive patients with undetectable plasma viral loads. 27 This finding was corroborated in further work showing that HIV envelope sequences were not significantly different between cells derived from GALT and PBMCs; however, the authors did not find evidence of HIV evolution in GALT. 28
In addition, recent phylogenetic work has shown that viral sequences derived from PBMCs were phylogenetic offspring from sequences derived from lymph nodes in patients with undetectable plasma viral loads. 29 Though this study was not performed in GALT, it demonstrates that focal HIV replication in lymphoid tissue can maintain PBMC infection during suppressive therapy. Together, these data support persistent HIV replication within the GALT, and that this local replication can maintain infection in the plasma through free movement of infected cells or virions between these compartments.
Despite these results, several investigations suggest that GALT is not the sole source of rebound viremia on treatment cessation. For example, a cross-sectional evaluation of multiple T cell subsets by McBride et al. showed that <20% of total HIV DNA was found in memory T cells with gut migratory capacity. 30 Further, sequence analysis of rebound plasma virus from three HIV-infected patients who experienced treatment interruption demonstrated that the postinterruption viral sequences were not GALT derived, suggesting an alternative source of rebound viremia. 31 This hypothesis is supported by a recent study that sampled GALT and lymph nodes before and after treatment interruption and found that rebounding HIV variants likely arise from many anatomic sites, rather than from a small viral population from a single location (e.g., GALT). 32
Nonetheless, the studies discussed earlier provide convincing evidence of persistent HIV replication in GALT, although the contribution of this replication to viral rebound remains unclear. Clinical observations are supported by genetic analyses, which indicate that viral gene expression in GALT is maintained even in the setting of undetectable plasma viral loads, and that there is likely cross-talk between plasma and tissue that may propagate infection in PBMCs. Though direct evidence is still needed (since attempts at isolation of replication competent virus from durably suppressed patients have failed 33 ), these data provide a foundation for exploring the mechanisms of HIV persistence in this tissue compartment.
Consequences of HIV GALT Persistence
Immune dysregulation in acute infection
As an early site of HIV exposure and infection, GALT plays an important role in the pathogenesis of HIV infection. The interplay between immune cell depletion and activation, local viral dynamics, and systemic immune dysregulation is complex and is the focus of thorough reviews. 34,35 It was observed early in the HIV epidemic that infection resulted in severe GI complications and was associated with increased mortality in AIDS patients. 36,37 This was caused by severe depletion of specific T cell populations within GALT that occur rapidly after infection (27% reduction within 4 weeks). 19
Specifically, IL-17-expressing T helper cell (Th17) populations were found to be preferentially depleted in simian immunodeficiency virus infection, which may lead to decreased immune function and disruption of gut epithelial integrity. 38 Studies in macaques and humans have shown these T cell decreases to occur within the first week of infection. 17 –19 The importance of T cell depletion within GALT has been supported by studies showing that elite controllers do not experience specific depletion of Th17 cells, suggesting that this immune dysregulation is a main driver of disease progression. 39
Delayed immune reconstitution
Persistent replication within GALT also represents a large obstacle to restoring immune function in HIV-positive patients. It is well documented that immune reconstitution is delayed in GALT after the initiation of cART, 19 and that a systemic inflammatory state is maintained even after plasma viral loads are undetectable. 40 This persistent inflammation, likely driven by continued GALT disruption, has been associated with poor outcomes and the development of comorbid conditions in these patients. 41 The mechanisms that are responsible for this delay have not been fully determined, but several studies have implicated ongoing CD8+ T cell activation, fibrosis, or impaired mucosal homing as contributing factors. 22,42,43
Importantly, ongoing viral replication in GALT could help explain this delay in immune recovery regardless of the specific mechanism(s) involved. Persistent exposure of viral antigen to antigen-presenting cells in the gut can increase inflammatory markers, 44 decrease epithelial integrity, 45 and increase microbial translocation, 46 continuing the cycle of local immune dysregulation and systemic inflammation. Microbial translocation is of particular concern, as this is associated with an increased incidence of comorbidities and hastened disease progression. 47,48 The profound immune depletion in GALT, with subsequent clinical complications, underscores the importance of eradicating residual viral replication in this compartment.
Challenges to Eradicating HIV GALT Replication
As introduced earlier, there are a number of factors that make GALT an ideal reservoir for HIV. The first line of defense against cART lies in the anatomy and location of GALT. Blood perfusion to the GALT via the extensive mesenteric arterial network is more variable than perfusion to other sites, and it may be reduced in situations where blood is needed in other organs (e.g., skeletal muscle during exercise, brain during shock). Further, the proximity of GALT to the colorectum, a site of HIV exposure, and the high density of target cells make it a site of initial infection and viral propagation.
Clinical evidence suggests that the HIV reservoir is established within days after infection, 49 and it is reasonable to expect that this may happen in GALT. Clinical studies show that the initiation of cART during the acute phase reduces, but does not eliminate, the HIV reservoir, 49,50 which is likely due to the very early establishment of the latent reservoir before cART can be initiated. Thus, alternative strategies for HIV elimination are required.
A critical obstacle to achieving HIV eradication in GALT is the potential for inadequate ARV distribution into this compartment. ARV penetration into gut tissues is highly variable, both between and within drug classes, 51 and is not easily predicted based on chemical structure or standard pharmacokinetic properties. 52 The lack of complete immune restoration in GALT after cART administration and the isolation of HIV RNA from tissues in patients with suppressed plasma HIV RNA levels provide indirect evidence that ARVs may not achieve adequate concentrations in certain areas of GALT. This is supported by tissue homogenate data showing that GI exposures of ARVs such as dolutegravir are 83% lower than plasma. 53
In addition, studies evaluating the utility of intensified ARV dosing regimens on the size of the viral reservoir have shown little effect, suggesting that anatomic or pharmacokinetic barriers may exist that prevent ARV penetration into GALT. 54,55 A study by Fletcher et al. compared ARV concentrations between PBMCs and mononuclear cells (MNCs) isolated from the lymph nodes, ileum, and rectum. 56 It was found that ARV concentrations in gut MNCs were a maximum of 100% lower than PBMC concentrations, and that lower concentrations correlated with slower HIV decay rate in these tissues. These data suggest that ARVs retain their efficacy provided they are able to reach an as-yet unidentified threshold concentration in target cells. However, the interplay between active replication secondary to reduced ARV exposure and latent infection remains unknown, as does the relative contribution of each of these processes to HIV persistence.
Despite the direct and indirect evidence that a lack of ARV tissue penetration contributes to HIV persistence, this idea remains controversial. The most frequently cited counterargument is the lack of observed drug resistance that would be expected to develop in the setting of subtherapeutic ARV exposure. 57 Given that resistance is known to develop rapidly when ARV concentrations drop below therapeutic concentrations in plasma, it is reasonable to question this pharmacologic assertion. In the absence of widespread resistance developing in patients, some have interpreted the continued detection of HIV gene expression as simply random egress from latency, having little to do with drug penetration. 57
However, it has recently been suggested that ARV exposure in tissue reservoirs may be so low that the threshold for resistance development is never met. 29 In other words, minimal ARV exposure allows for continued replication of wild-type virus, which can outcompete resistance variants that could otherwise emerge. In this way, inadequate ARV penetration into tissues may contribute to ongoing replication that is congruent with clinical observations of low-level viremia in patients receiving cART.
In addition to propagating immune dysfunction and systemic inflammation, inadequate ARV penetration into target cells may also hinder efforts of latency reversal and blunt the corresponding host immune response. 22 Reversal of HIV latency in quiescent memory T cells using small-molecule drugs represents a promising approach for reduction or eradication of the HIV reservoir, and several drug candidates are currently undergoing clinical evaluation. 58,59
A key assumption underlying these efforts is that virions produced from these re-activated T cells will be unable to infect nearby healthy cells and stimulate new rounds of HIV replication. However, this assumption is valid only if ARVs are present at sufficient concentrations to inhibit viral replication. However, if enough healthy T cells remain unexposed to ARVs 56 in the setting of latency reversal, the reactivation of quiescent cells could result in no effect, or possibly an increase in the size of the viral reservoir through new rounds of infection.
There are several barriers that complicate the optimization of ARV therapy for HIV eradication in GALT. First and foremost is the limited knowledge of the specific factors that influence ARV disposition in GALT. ARV penetration into tissues is variable and dependent on several physicochemical and pharmacokinetic characteristics. 51 Volume of distribution and plasma protein binding are important variables and vary widely from drug to drug. Drug transporters and metabolizing enzymes are also known to affect the absorption and distribution of ARVs, and there are many examples of clinically significant drug-drug interactions involving these proteins. 60 Though the specific transporters or enzymes utilized by ARVs vary between drug classes, the metabolizing enzymes of the CYP450 family, particularly CYP3A4, and the efflux transporters p-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and the multi-drug resistance proteins (MRP) are responsible for a significant portion of ARV transport and metabolism. 61
As such, alterations in the expression and/or activity of these proteins can have a dramatic effect on local ARV concentrations. As an example, the penetration of atazanavir (ATV) into the brain was increased by 5.4-fold in P-gp/BCRP knockout mice compared with wild-type mice, showing that a lack of functional efflux transporters at the blood-brain barrier allows more drugs to enter the tissue. 62 Other groups have shown that the expression of the uptake transporter family OAT1 at this site also modulates tenofovir brain penetration. 63
These findings have been corroborated in other tissues such as the testes, where it was shown that the inhibition of P-gp and BCRP in human Sertoli cells increases the testicular uptake of ATV, 64 and in P-gp/BCRP knockout mice, where ATV concentrations in testicular tissues were increased by 4.6-fold compared with wild-type mice. 62 A recent paper by Huang showed that the penetration of protease inhibitors into human testes was variable and may be dependent on drug transporter expression and localization. 65
Expression and activity of these proteins is variable along the GI tract. 66,67 For example, protein expression of P-gp and BCRP was found to be threefold higher in the ileum versus the colorectum, with the opposite being true for the efflux transporter MRP2 (1.5-fold lower in ileum vs. colorectum). 67 CYP3A4, an enzyme responsible for the metabolism of protease inhibitors, has been shown to be expressed at varying levels in GI tissue, and it is highest in the duodenum. 66,68 In combination, these factors may explain the large differences observed in ARV exposure in tissues, including the GI tract.
For example, the exposure of the P-gp substrate raltegravir in the splenic flexure was shown to be 2.8-fold higher than the terminal ileum or colorectum. 69 This is inversely related to the expression patterns of P-gp in the GI tract, 67 suggesting that decreased RAL efflux out of enterocytes secondary to focal decreases in efflux transporter expression can result in compartment-specific increases in RAL exposure along the GI tract. This complicates attempts to achieve maximum drug exposure in the tissue, as the local expression of drug transporters and enzymes may be the primary driver of tissue concentration.
To date, there has been no evaluation of the relationship between these variables and ARV GALT exposure; however, given the numerous examples of altered plasma pharmacokinetics secondary to drug transporter or metabolizing enzyme interactions in the gut, it is reasonable to expect that changes in these variables will also have an effect on local ARV exposure in GALT.
An additional barrier to ARV optimization in GALT is the lack of target concentrations for efficacy in this setting. Plasma target concentrations are well established and are easily achievable by standard dosing regimens (derived from early dose finding studies), but similar target concentration values for eradication of residual HIV replication in tissues have not been determined. HIV RNA has been detected in the GI tract of patients receiving cART who had detectable ARV concentrations in these same tissues, providing evidence that current regimens are not sufficient to stop residual replication, although these patients had only been receiving cART for several months. 20 The lack of efficacy observed in ARV intensification studies 54,55 shows that more sophisticated methods of determining effective concentrations are needed to define pharmacokinetic targets in this compartment, as higher concentrations may be needed for a longer period of time in tissues compared with plasma.
In a study of pre- and post-cART reverse transcriptase (RT) sequences in eight HIV-infected participants, it was found that sequence diversity decreased 1.5- to 2-fold throughout the gut during treatment, and that zidovudine (ZDV)-resistant variants were concentrated in the gut versus PBMCs, demonstrating that efficacy targets may be different between these compartments. 70 The ability to define exposure-response relationships in GALT and other tissue reservoirs, as has been done in the female genital tract for HIV prevention efforts, 71 will be critical to overcoming HIV replication in this compartment. Defining these relationships is unlikely to change current dosing strategies (though dose intensification studies suggests that this would have little effect); however, a well-defined PK target in tissues would inform the development, or selection, of targeted therapies to stop replication.
The challenges described earlier highlight the difficulty in eliminating HIV persistence in GALT. Although some barriers are unavoidable (e.g., high target cell concentration, size and complexity of GALT, etc.), others, such as inadequate drug penetration or the contribution of drug transporters or metabolizing enzymes, can be acted on in a rational way to improve therapeutic success in this area. The next section focuses on ways that clinical pharmacology approaches can inform the design of targeted therapies for HIV eradication in GALT.
Opportunities for Pharmacologic Intervention
Emerging technologies to understand ARV distribution
The ability to accurately quantify ARV concentrations in tissue reservoirs such as GALT is critically important in understanding the distribution of these drugs. To date, most evaluations of ARV tissue concentrations have used liquid chromatography-mass spectrometry (LC-MS) analysis of tissue homogenates. 51 Though traditional LC-MS methodology can provide clinically useful quantitative data, it does not have the ability to identify distributional patterns within the tissue, as the entire sample is consumed in the homogenization process. This is a critical limitation of the technology, as it has been shown for other drugs that distribution across tissue is not uniform. 72
For example, if the majority of a particular ARV were located in a specific tissue compartment (e.g., epithelial layer) with no distribution into the rest of the tissue, LC-MS may overestimate the penetrative ability of this ARV. This is particularly concerning if HIV replication is occurring in focal areas within a tissue that cannot be reached by ARVs. Further, because the majority of ARVs have intracellular sites of action, concentrations within cells are the largest determinants of antiviral efficacy. MNC isolation from reservoir tissues overcomes the limitations of LC-MS by reporting ARV concentrations only in the cells of interest. 56 However, this method cannot easily or completely account for drug lost during the isolation process, which can significantly underestimate true intracellular concentrations, 73 nor can it distinguish cell populations derived from different areas within a tissue. Therefore, it is crucial to use alternative technologies to define ARV distribution into tissues and cells.
Mass spectrometry imaging (MSI) is a growing field that has numerous potential biological applications and is already being implemented in the drug development process. 74 MSI combines the sensitivity and specificity of LC-MS with the ability to visually observe distributional patterns within tissues. Using infrared matrix-assisted laser desorption/electrospray ionization (IR-MALDESI), a novel MSI method, we have demonstrated that the distribution of ARVs such as efavirenz is heterogeneous within the colorectum, concentrating in the mucosa and lamina propria and corresponding to areas of high CD3+ T cell density. 75
Tissue drug imaging can be particularly powerful when combined with other techniques such as in situ hybridization (ISH) or immunohistochemistry (IHC). Leveraging all of these methods in combination, it is possible to compare ARV distribution to that of HIV target cells (IHC) or to HIV itself (ISH) to determine whether ARV localization corresponds with areas of local HIV replication. 76 Because IR-MALDESI is quantitative, 76 –78 it is also possible to determine what local concentrations are needed within a tissue compartment to effectively suppress HIV replication, aiding in the search for target concentrations in tissues.
Raman spectroscopy (RS) is another promising imaging technique that has the potential to provide ARV distributional data within unperturbed tissues such as GALT. Utilizing the principles of Raman scattering and confocal microscopy, RS can identify exogenous substances, such as small-molecule ARVs, in heterogeneous tissue matrices. 79 Similar to IR-MALDESI, RS can potentially identify areas of ARV localization. Unlike IR-MALDESI, however, RS is able to temporally analyze small-molecule distribution, providing spatial information in real time. 80 Recently, RS has been combined with optical coherence tomography technology to identify tenofovir distribution in mucosal tissues at various tissue depths. 81
Another advantage of RS, and one that is shared with IR-MALDESI, is the high spatial resolution that these technologies can provide. Both technologies have been demonstrated to accurately detect drugs at resolutions of 100 microns or lower. 79,82 This is especially important for distinguishing ARV distribution among different cell populations in heterogeneous tissues such as GALT. As these technologies improve, it may be possible to distinguish ARV concentrations at different cellular locations, 83 providing even more specific PK information in the cytosol (relevant for most ARVs) versus the nucleus (relevant for integrase inhibitors).
Droplet microfluidics is a pioneering technology that allows for high-throughput, single-cell assays. 84 In the case of HIV persistence, where proviral DNA may be found in a relatively small number of cells, understanding how the exposure of a current or novel therapy exerts its pharmacodynamic effect ex vivo would provide a foundation for the optimization of therapy. By encapsulating single cells inside small-volume (1 pL) droplets and performing reactions within each droplet, microfluidics overcomes the disadvantages of current techniques. 85
This technology was previously used to perform cellular screening assays for drug effect and genetic analysis to link genotype to phenotype in a single-cell population. 86,87 Though this technique has not yet been used to evaluate small-molecule concentrations, it shows promise in the area of cellular ARV PK, as differential exposure between and among cell types can be evaluated with a high frequency (2,000 Hz), with simultaneous evaluation of PD effects (protein or gene expression changes).
Evaluation of small-molecule tissue and cellular distribution using novel technologies is a burgeoning field. IR-MALDESI and RS have a growing body of evidence demonstrating their utility in this arena. Microfluidics shows additional promise for performing cellular PK/PD evaluations. The advantages and disadvantages of each of these technologies are summarized in Table 1. One limitation that is shared by all of these methods is the difficulty in accounting for synergy among ARVs, which may result in adequate control of replication despite suboptimal exposure of the individual drugs. In addition, though IR-MALDESI has been used to evaluate ARVs from multiple classes, 88 RS and microfluidics have had limited or no use in ARV pharmacology to date and it is unknown whether or not these methods will have widespread utility. However, exploring the use of these techniques and others to evaluate ARV distribution in GALT is an important step toward HIV eradication.
LC-MS, liquid chromatography-mass spectrometry.
Defining the factors influencing ARV disposition
When viewed in isolation, drug exposure in tissues is limited in its ability to inform the development of HIV therapies that are targeted at GALT or other tissue reservoirs. Beyond simply defining and describing the problem (e.g., a certain drug is sequestered in the gut mucosa, allowing for ongoing HIV RNA expression in submucosal T cells), distributional data need to be evaluated from the perspective of improving ARV exposure. As stated earlier, ARV disposition in the GALT is dependent on many factors affecting absorption and distribution (Fig. 1). The physicochemical characteristics of the ARVs themselves likely play an important role in tissue exposure. It has already been shown, for example, that drugs with low plasma protein binding penetrate better into mucosal tissues. 52
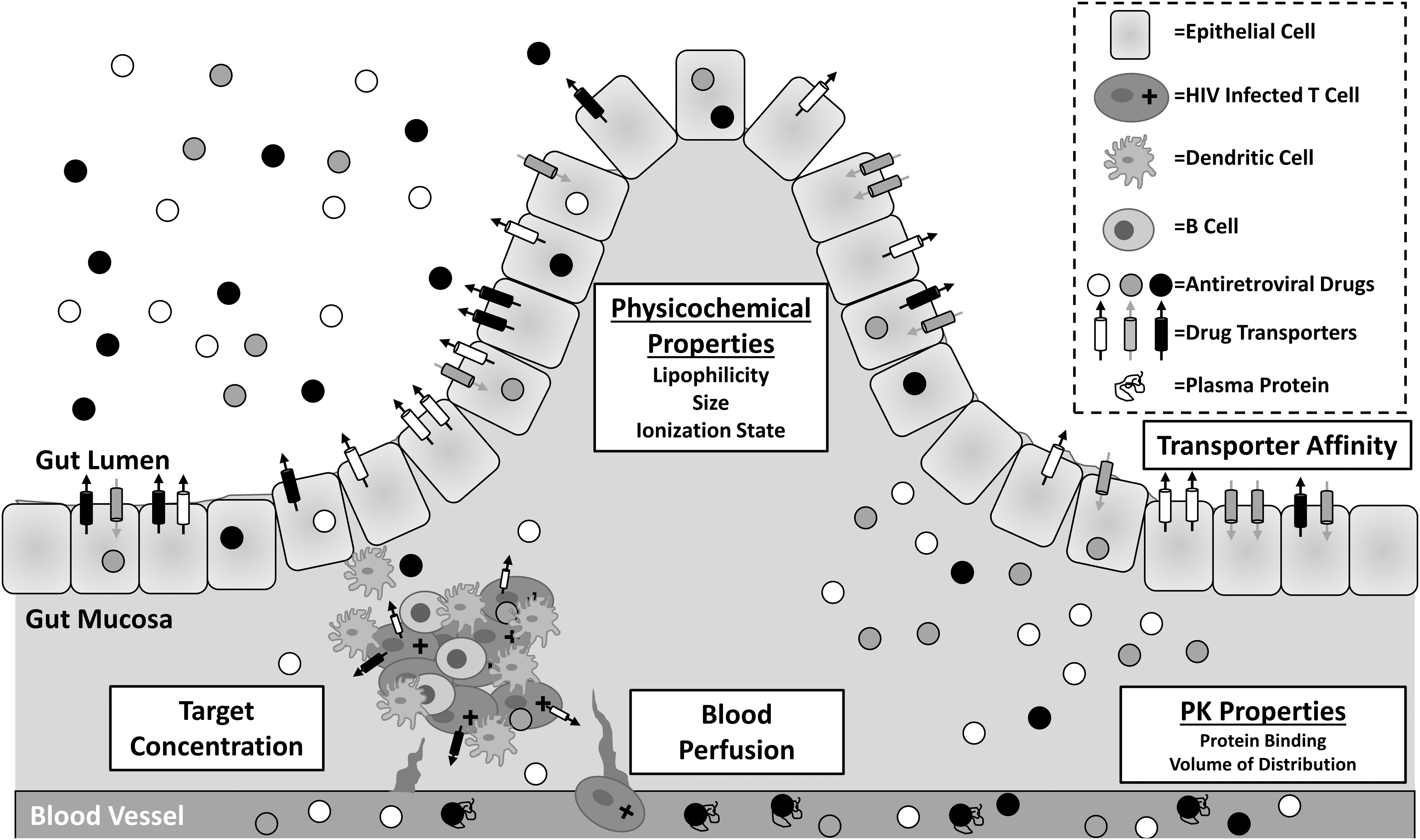
Factors influencing antiretroviral disposition and efficacy in GALT. Solubilized drugs (white, gray, and black circles) can penetrate into the intestinal tissue from peripheral blood supply or directly from the intestinal lumen. Penetration arising from the blood is dependent on the amount of local perfusion as well as on pharmacokinetic properties of the drug, that is, drug bound to plasma protein (shown within blood vessel) cannot enter the tissue. Penetration from the gut lumen is dependent on physicochemical properties of each drug as well as on the affinity for drug efflux (white, black) or uptake (gray) transporters on the surface of epithelial cells. GALT-specific exposure is also dependent on local blood perfusion and drug transporter expression on the surface of lymphocytes. GALT, gut-associated lymphoid tissues; PK, pharmacokinetic.
Further, the retention time of these drugs (as a function of their half-life in tissues) is also critical, as it has been shown that ARVs such as indinavir are rapidly cleared from the lymph compartment, despite achieving equilibrium with plasma concentrations. 89 In addition, there may be certain components of the molecular structure of ARVs that favor increased GALT exposure, and these features can potentially be utilized in the development of nanoformulations that are targeted at tissue reservoirs. 90,91
Targeted nanoformulations have shown success in cancer therapeutics, and several nanoformulated drugs are approved for use in breast and pancreatic cancer, achieving increased exposure into tumors and increasing survival rates. 92 Among ARVs, a nanoformulated version of indinavir was shown to achieve a sixfold increase in lymph node concentrations and greater exposure in plasma. 93 Additional formulations for drugs such as abacavir, rilpivirine, and efavirenz are already in development, masking unfavorable PK characteristics to maximize ARV tissue exposure. 94 –96 For example, a dimeric prodrug formulation of abacavir, which functions as a P-gp inhibitor when dimerized but that can still exert anti-HIV activity when cleaved to its monomer form, has been developed to increase abacavir penetration into the central nervous system. 94 Similar approaches for other ARVs to increase GALT exposure should be considered.
Another important step toward eradication is to identify which drug transporters or metabolic enzymes either adversely or favorably affect ARV exposure in the GALT. Exploiting these factors to increase ARV GALT penetration from the lumen (e.g., P-gp inhibition on enterocytes to decrease efflux) or systemic circulation (e.g., CYP3A4 inhibition in the liver to increase half-life) may overcome the inherent challenges in treating GALT persistence. Several studies have shown drug transporter expression to be inconsistent along the GI tract, 67 but transporter localization in relation to GALT has not been evaluated in this context. A potential explanation for this gap in knowledge is the lack of consistency with which transporter expression is evaluated and reported, as the methodology used in these studies is highly variable, with groups using quantitative polymerase chain reaction, immunoblot, LC-MS proteomics, IHC, or some combination of these methods. 97 –100
Further, there has been no formal analysis of the agreement of these techniques with one another. Gene expression has been shown to vary drastically from end-protein expression, and even measures of transporter proteins may give very different results. 100 To fully characterize which drug transporters or metabolic enzymes affect local ARV concentrations in GALT, a consensus should be reached on the best expression measures in tissues. Studies formally comparing these methods with each other and examining which method of measurement provides the most useful comparisons to ARV distribution are greatly needed.
Leveraging ARV distribution data against the biologic factors that influence these distribution patterns will provide the most informative data for the design of novel therapies that are targeted at tissue reservoirs, including GALT. In addition to identifying which ARVs achieve the highest exposure in GALT and whether the exposure occurs where latent and/or reactivated HIV virus is located is critical. Further, defining which drug characteristics, transporters, or enzymes should be targeted or avoided when developing new therapies or optimizing existing ones would refine novel strategies to be pursued. Importantly, defining the characteristics that affect ARV exposure in GALT is likely to also inform the exposure of latency reversing agents and other small-molecule compounds in GALT. Although specific factors may differ between drug classes, the lessons learned from the evaluation of ARVs in this tissue can be easily translated to LRAs, for which GALT exposure is equally important.
Utilizing preclinical models to streamline drug development
The location and nature of many active HIV reservoirs make them difficult to sample in human participants. Limited repeat sampling can be accomplished in some sites, including GALT, but extensive evaluation of human GALT tissue remains a challenge, particularly for the ability to recover virus from tissue biopsies. These limitations necessitate the use of preclinical animal models to study HIV persistence. Unfortunately, it has been shown that ARV distribution can be variable between species, with tissue ARV penetration varying greatly between humanized mice, nonhuman primates, and humans. This is especially problematic when plasma data from animal models are used to select effective doses for humans, potentially adversely affecting clinical trials.
The reasons for these discrepancies are multifactorial, but species differences in drug transport and metabolism are likely contributors. An extensive review by Lin summarizes some of these differences, including that the concentrations of CYP3A and CYP2C are increased by 1.8- and 12-fold in rats compared with humans, 101 which can lead to an increased metabolism of substrates (e.g., protease inhibitors, NNRTIs) and decreased plasma or tissue exposure over time. Further, our group has shown substantial differences in gene and/or protein expression of several drug transporters between humanized mice and nonhuman primate models, which may affect local concentrations of ARVs that are transported be these proteins, particularly those affected by P-gp (e.g., abacavir) and BCRP (e.g., dolutegravir). 100,102
The challenges in reconciling data from preclinical species as they relate to HIV persistence have been recognized by the field. 103 However, it is obvious that an animal model that accurately reflects ARV tissue distribution in humans or that can be accurately scaled to predict human distribution would be helpful in the evaluation of candidate ARVs for targeting GALT. The generation of reliable interspecies scaling factors for ARV distribution and metabolism would streamline ARV development when tissue targeting is required. To accomplish this, studies evaluating ARV distribution and metabolism across multiple species, including humans, and particularly focusing on GALT, should be conducted. These data can form the foundation for quantifying the variability of ARV distribution across models and developing models to accurately predict these endpoints in humans, and greatly inform eradication strategies, including anticipated combination strategies in which drug-drug interactions are possible.
Conclusion
HIV persistence represents the largest obstacle in the search for a cure. If ongoing replication in GALT is to be eliminated, an understanding of how this reservoir propagates is needed. Although this compartment presents unique challenges to eliminating replication, the pharmacologic tools described here offer promising solutions. Tissue imaging provides distributional data that have been previously unattainable, and it will allow the field to assess whether or not ARVs have a significant presence in reservoirs by identifying and quantifying within-tissue localization.
This has enormous implications for HIV persistence; heterogeneous ARV distribution in the GALT would provide convincing evidence that current therapies are simply not present where they are needed, whereas reproducibly broad distributional patterns would suggest alternative reasons for persistent infection. Demonstrating that HIV replication is occurring in areas or cells with low ARV exposure would provide further evidence that inadequate ARV tissue exposure contributes to HIV persistence.
In addition to clarifying the mechanisms driving HIV persistence in GALT, the tools of pharmacology can be used to inform the development of targeted therapies for HIV eradication. Defining the factors governing ARV exposure in GALT will help to identify which variables to target or avoid during the development of novel small molecules, particularly once preferred methodologies for measuring these factors are identified. In addition, performing these evaluations in multiple preclinical species will provide foundational knowledge of interspecies tissue distribution differences. This will be critical for the evaluation of candidate ARVs and latency reversing agents for targeting reservoirs by preventing the inappropriate interpretation of data from one species to another. Ultimately, these data will allow for a more directed design of therapies that are aimed at eradicating HIV reservoirs and potentially curing HIV.
Footnotes
Acknowledgments
The authors are grateful for the National Institutes of Health Grant R01 AI111891 (ADMK, CT), Martin Delaney Collaboratory Grant U19 AI096113 (ADMK), and the American Foundation for Pharmaceutical Education PreDoctoral Fellowship (CT).
Author Disclosure Statement
No competing financial interests exist.