
Abstract
Mother-to-child transmission (MTCT) of HIV offers a good opportunity to study the dynamics of early viral evolution in the host environment to which the virus has partially adapted. Such studies would throw light on the unique features of the infecting viruses, which will subsequently help to design preventive or therapeutic measures against the newly infecting and evolving strains of HIV. Therefore, we undertook a study to determine the genetic divergence of proviral envelope sequences from the HIV-infected infants (<2 years). Detailed analysis revealed unique features of potential N-linked glycosylation sites (PNGS) and their frequency of occurrence that built on the difference in length of the V1V2 region of the envelope sequences. Surprisingly, frequency of PNGS in the V5 region was found to revert rapidly, in about 75% of the sequences, which could surmise a fitness disadvantage in the variant forms. Further, a stable net charge was observed in the V2 and V3 regions prompting us to speculate on the established interaction of the transmitted variant with the integrin α4β7 receptor and R5 co-receptor, respectively. In brief, our observations suggest that differences in the length of the variable regions and variation in the frequency of PNGS in the envelope of the viruses obtained from very recently infected individuals in our population could be important characteristics of the unique quasispecies that is responsible for the spread of HIV in the early stages of infection in MTCT.
Introduction
A
During the event of HIV-1 transmission, the virus evades the gatekeepers by means of antigenic variation resulting from an extraordinary degree of genetic diversity, conformational flexibility, and extensive glycosylation of the HIV-1 envelope surface. 2 Therefore, solving the challenging scientific puzzle of identifying the “gatekeeping” mechanisms in the early events of HIV-1 transmission is critical for the development of effective HIV-1-preventive measures. 3 Novel experimental approaches that generate strong knowledge on the functional features of the HIV-1 virus variants are also needed to find a solution to this problem. In practice, it has been impossible to identify and characterize the virus that is responsible for establishing infection, at or near the moment of transmission. 4 Most of the genotypic and phenotypic studies in both horizontal and vertical transmission in recently infected individuals are characterized by the presence of highly homogeneous virus 5 while, heterogeneous virus populations with enormous genetic diversity are seen in the later stages of infection. 5
Within a few weeks of infection, envelope-specific antibodies are identified in individuals, 6 but these fail to neutralize the virus. However, these non neutralizing antibodies have the ability to activate the complement cascade that can result in both viral inactivation and enhanced infection. In contrast, development of neutralizing antibodies takes several weeks to months after acute infection. 7 These antibodies usually target the immunodominant and variable regions of the virus. 8 Ideally an effective vaccine should elicit a neutralizing antibody response of great breadth and potency, and be capable of overcoming the genetic variability, structural complexity, and neutralization escape mechanisms of the virus. 9 Hence, it is of fundamental importance to identify antibodies that neutralize a wide spectrum of globally circulating viral isolates and their potential targets for HIV-1 immunogen design. 8
The primary viral envelope component, the gp120 spike, composed of five variable regions (V1–V5) and five constant regions (C1–C5), exhibits a relatively flexible structure. Despite being functionally important for forming a complex with the CD4 receptor on the host cell, 10 these molecules are able to accommodate high levels of variability and are the foremost targets for neutralizing antibodies. In particular, the variable regions are reported to: (1) protect the CD4 binding-site from neutralizing antibodies and play a role in infectivity and tropism (V1 region), 11 (2) bind the Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) (V2 region), 12 (3) modulate neutralization sensitivity by hiding conserved epitopes (V1 and V2 regions together), 13 (4) bind both CCR5 and CXCR4 cellular co-receptors (V3 region), 14 and (5) influence Env conformation and glycan packing (V4 and V5 regions). 15 In addition, frequency of occurrence and pattern of N-glycosylation in gp120 can impact protein folding, 16 intracellular transport, 17 co-receptor binding, 18 immune dominant domains, 17 and may play a major role in viral transmission 19 by influencing infectivity fitness. 20 By changing the lengths and sequences of the variable loops and by modifying the number and position of attached glycans during the course of infection, HIV-1 creates an ever evolving glycan shield, constantly generating novel env variants resistant to the concurrent array of host antibodies.
An understanding of the conformational changes in gp120 and gp41, and its mechanisms and kinetics of binding with receptors and co-receptors during transmission would prove useful for establishing an algorithm to boost host immunity against the evolving strains of HIV. Hence, we undertook this study on the molecular characterization of the envelope of HIV-1 variants transmitted recently through the event of MTCT.
Materials and Methods
Sample collection
Blood samples were collected prospectively from infants who acquired infection from the mother through the vertical route, either during the event of pregnancy in utero, at birth, or postnatally through breast milk, and were diagnosed to be HIV positive during the first few weeks of birth. Due to insufficient sample availability, CD4 count and viral load could not be tested on these samples. The demographic details of the children are given in Table 1. The chosen sample size was 20 and the sampling period was from 2013 to 2014. Ethical clearance for the study was obtained from the Institutional Ethics Committee.
F, female; M, male.
Age/sex of the infant at the time of sample collection.
DNA isolation and envelope amplification
Proviral DNA was extracted directly from whole blood using the QIAamp blood kit (Qiagen GmbH, Hilden, Germany) following the manufacturer's instructions. DNA was estimated and quality checked using NanoDrop (Thermo Scientific, DE) and stored at −80°C until use. Full-length env (gp160) gene was amplified by nested polymerase chain reaction (PCR). First-round PCR was performed with 2 × kapa HiFi hotstart ready mix (Kapa biosystems), 10 mM of each deoxy nucleotide triphosphate (dNTP), 0.5 mM of primers vif1, and ofm19 (Table 2) with 100 μg of DNA as template in a total reaction volume of 25 μl. PCR conditions for the first round were 95°C for 5 min, followed by 30 cycles of 98°C for 20 s, 62°C for 15 s, 72°C for 2.5 min, and with a final extension of 72°C for 5 min. One microliter of first round PCR product was used as template for the second round PCR, with identical cycling conditions and PCR mix except for the primers. Primers used in the second round were env In A and env In N (Table 2).
PCR purification and sequencing
Amplified PCR products were purified using Qiaquick PCR purification kit (Qiagen). Purified products were quantified and checked for purity by measuring absorbance at 260 and 280 nm. The amplicons were sequenced using a variety of internal sequence specific primers (Table 2). Three additional sequencing primers were used to fill up the gap in a few sequences. Sanger sequencing was performed using the 3100 Avant Genetic Analyzer (Applied Biosystems).
Sequence analysis
Full-length env contigs were edited based on the purity of the peaks in the electropherogram and assembled manually against an Indian reference sequence (pIndie-AB023804) using seqscape assembly program, version 2.5, with prescribed default parameters. Multiple sequence alignment was done with MEGA6 software using HXB2 as the reference sequence.
Quality of the sequences was checked with the Los Alamos National Laboratory database (
Phylogenetic analysis was carried out using Maximum Likelihood (ML) method with a poisson model. Phylogenetic tree was constructed using MEGA6 software with bootstrap values (100 replicates) for relevant nodes being reported on a representative tree. To construct the phylogenetic tree, HIV group M subtypes and circulating recombinant forms (CRFs) sequences from the different regions were downloaded from the Los Alamos database (
Potential N-linked glycosylation sites (PNGS) in the envelope were identified using the tool, N-GLYCOSYTE (
Results
Full-length HIV-1 subtype C envelope genes were successfully amplified and sequenced from 17/20 recently diagnosed HIV-positive infants (aged between 42 days and 15 months) who acquired infection via vertical transmission from HIV-positive mothers. The alignment of the 17 sequences is provided separately as Supplementary Fig. S1 (Supplementary Data are available online at
The ML phylogenetic tree constructed using MEGA software, version 6.06, based on the full-length env sequence, classified all variants that were sequenced in this study, within the HIV-1 clade C (Fig. 1). The tree showed that all the 17 isolates clustered tightly with the subtype C reference sequences. Within the subtype C cluster, majority of the study samples were found to be closely associated and intermixed with Indian and Chinese reference strains; South Africa and Brazil sequences were found to be on an adjacent branch. No recombinants were identified among the 17 sequences. In the phylogenetic tree, viral sequences from patients have been circled with different colors according to the ethnic background of the sample.

Molecular phylogenetic analysis of the envelope sequences of the 17 vertically transmitted viruses with HIV-1 group M subtype specific reference sequences downloaded from the Los Alamos database. HIV-1 subtype C sequences are represented as bubbles of different color. The color codes are defined on the left. The phylogenetic tree was constructed from amino acid alignments using Maximum Likelihood method based on the Poisson correction model. The tree was drawn to scale, with branch lengths measured as number of substitutions per site. No. of bootstrap replications was set at 100. Values less than ∼70 are not shown. Color image available online at
Co-receptor usage was predicted based on the V3 loop sequences. Figure 2 represents the diversity of amino acid sequences in the V3 region of the envelope. A net charge <+5 in the V3 region predicts CCR5 usage (R5 virus), whereas a net charge >+5 predicts CXCR4 usage (X4 virus). A net charge equal to +5 indicates R5 usage unless accompanied by the presence of arginine/lysine at position 11 and 25 in the V3 region. All except two isolates were predicted to use CCR5 as the co-receptor by at least two of the methods employed. Interestingly, the viral envelope obtained from these two samples (NIRT_ENV004 and NIRT_ENV017) also had glycine at position 13 in the V3 region. Further, the sequence, NIRT_ENV017 demonstrated considerable variability in the V3 region and possessed the GPGR motif. As a result, this sample was predicted as belonging to A/AG by the jpHMM tool.

Alignment of the V3 amino acid sequence of the 17 vertically transmitted HIV-1 subtype C isolates. The sequences were aligned with HXB2 reference sequence. The overall positive and negative charge, number of amino acids and residues at position 11 and 25 for each isolate are shown on the right. The V3 crown/tip is boxed in blue and red, representing GPGQ and GPGR, respectively. Dots and dashes indicate conservation and deletion/insertion of amino acids, respectively. Color image available online at
A comparative box plot analysis of the length of each variable region in the envelope of the vertically transmitted virus variants is represented in Figure 3. No significant difference in length was observed in the V3 region of the viral envelope; while the V3 region comprised of 35 amino acids in 16 samples, there were 34 amino acids in 1 variant. However, statistically significant broader length variability was detected in the length of the V1 and V5 regions, ranging from 18 to 33 amino acids and 4 to 11 amino acids in each of these regions, respectively. The V4 regions comprised of 27–31 amino acids in all variants, except 1 (NIRT_ENV001), which had only 21 amino acids. Among horizontally transmitted viral variants, the length of the V5 region is reported to be maintained more or less constant throughout the course of the disease. 22 In contrast, in acute infection through vertical transmission, dramatic changes in the length of the V5 region were observed between the viral isolates.
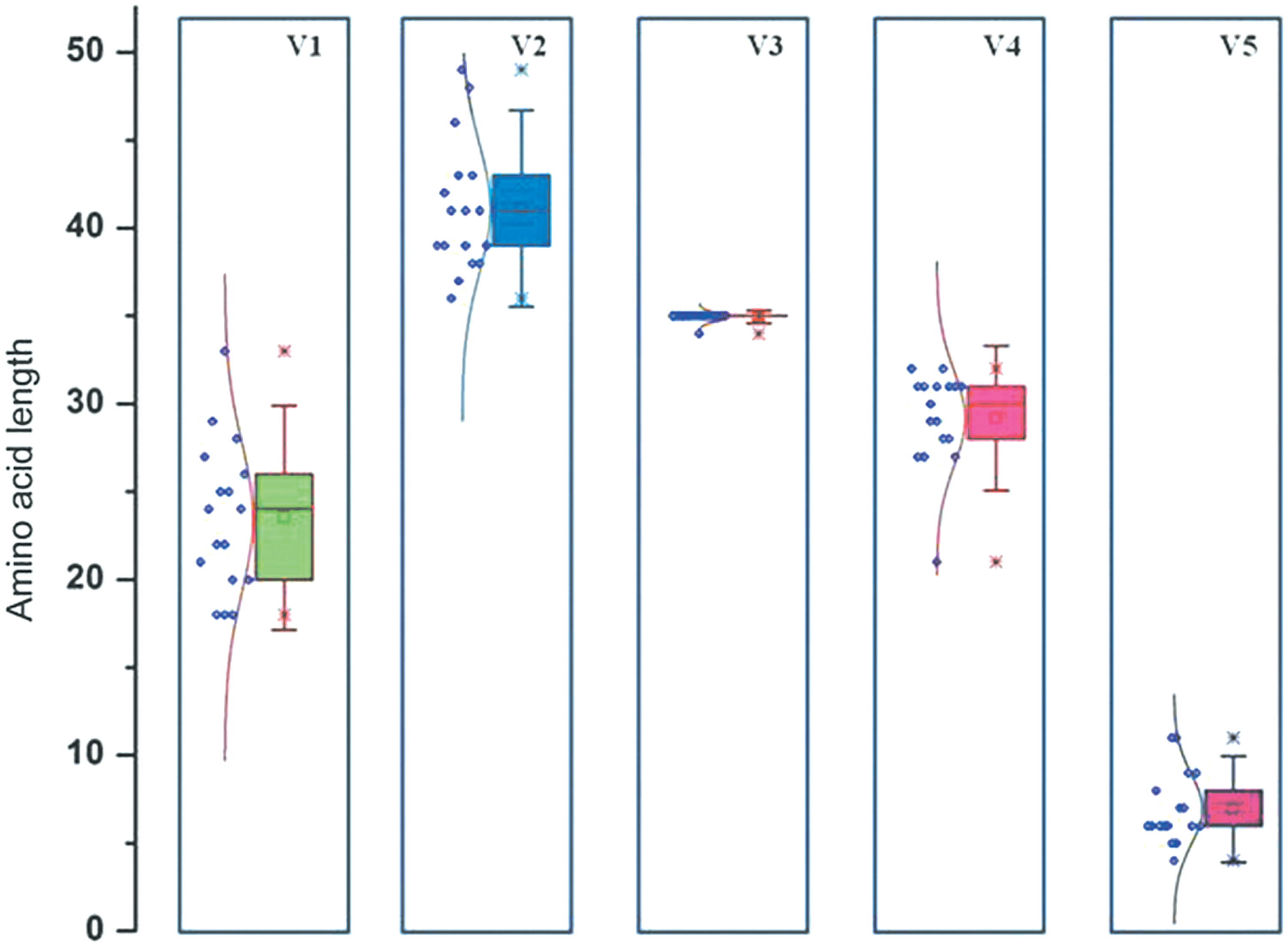
Comparative box plot analysis representing difference in length of each variable region in the experimental sequences. The box plot shows median, minimum, and maximum values. Each dot represents the number of amino acids in each sequence. Color image available online at
Difference in position of PNGS in the different env regions of the experimental sequences are shown in Figure 4. As can be seen from the figure, number of PNGS ranged from 27 to 34 in the sequences analyzed, with an average of 30 PNGS. The percentage of PNGS in the different variable regions was calculated and found to range between 31% and 48%, with an average of 38%. A significant difference in the number of PNGS (5–8) was identified in the V1/V2 region in the sequences obtained in this study. Interestingly, more than one PNGS was present in the V3 region of some of these sequences, which is in contrast to the single PNGS reported among the horizontally transmitted variants. Further, we observed deletion of PNGS in majority of the V5 sequences (13/17).
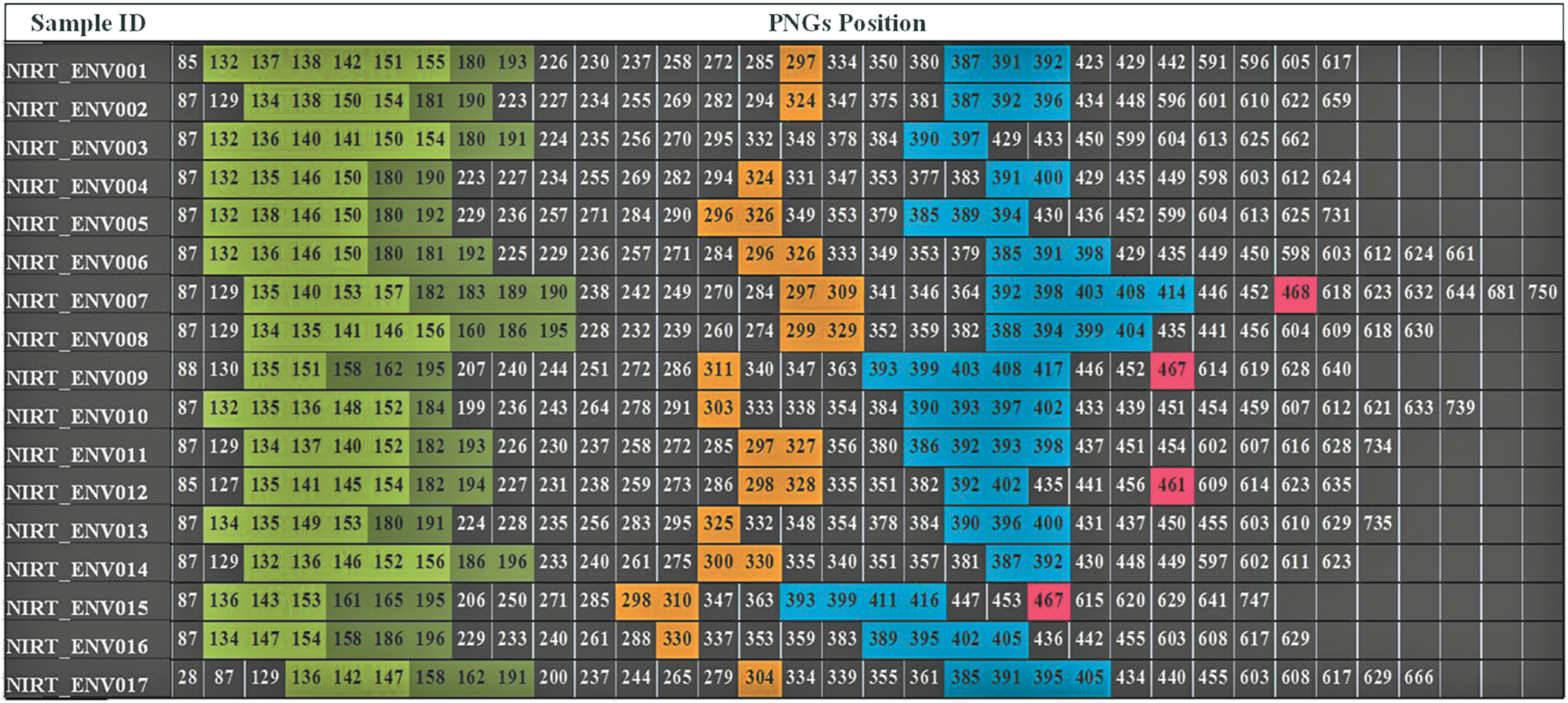
Position of PNGS in the env sequences. The numerical value represents the position of PNGS in each sequence. Gaps and deletions within the sequence were removed before the prediction of PNGS. Positions shaded with green, dark green, yellow, cyan and pink color denote deposition of PNGS in the variable region (V1–V5), respectively. Positions were defined with reference to the HXB2 sequence. PNGS, potential N-linked glycosylation sites. Color image available online at
The electric charge of each region (positive, negative, and net electric charge distributions) was analyzed to perceive the impact of immune pressure on the recently transmitted viral isolates (Fig. 5). Net electric charge of V2 and V3 regions was found to be higher in all the sequences analyzed. Transition of electric charge in the variable regions V1, V4, and V5 was observed in some of the env sequences. The net electric charge of the V1 region was comparatively lower than that of the V4 and V5 regions, which was characterized as neutral. However, when region-wise negative electric charges were considered separately, a statistically significant increase in the charge of the V2 region was noticed.
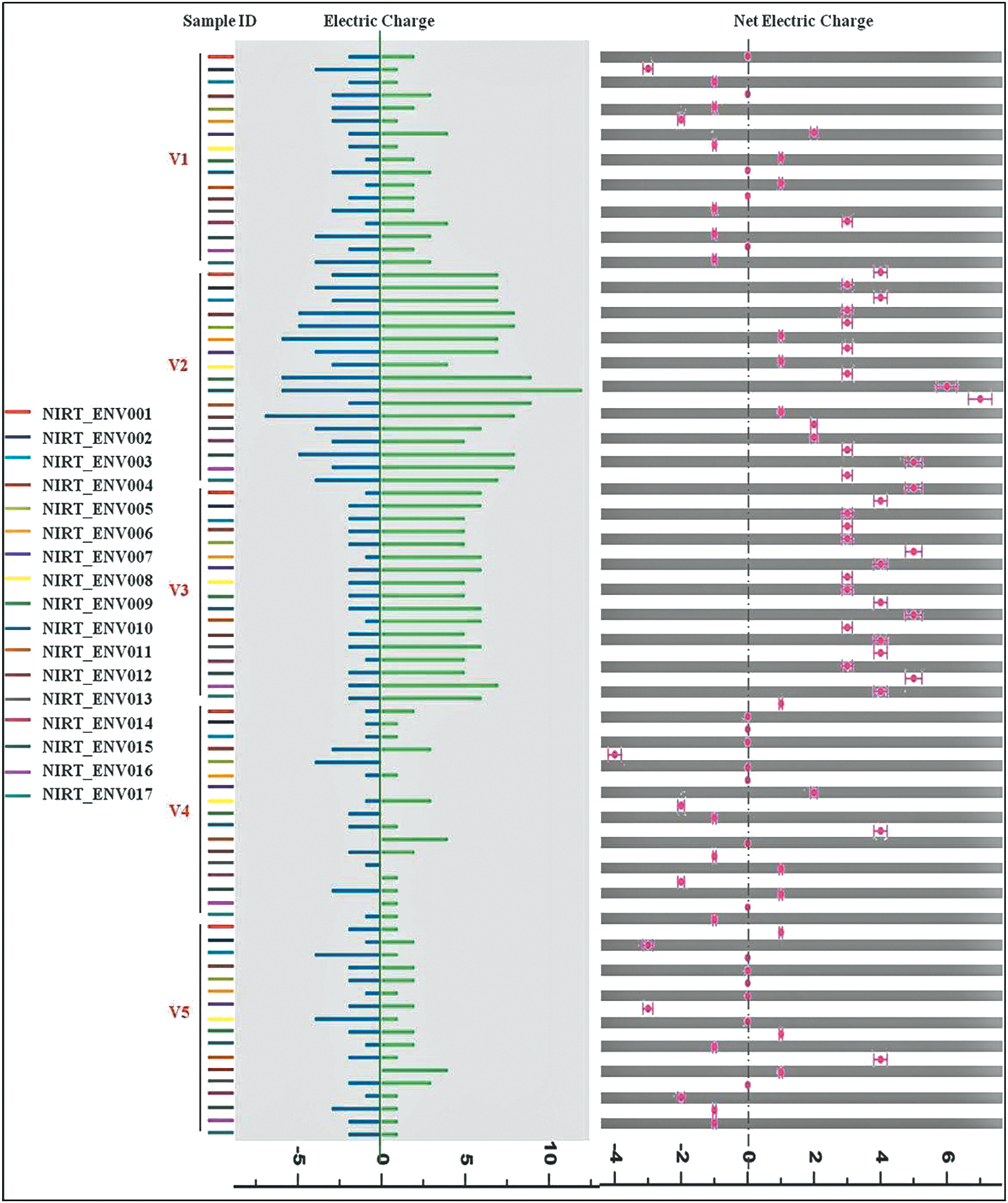
Electric charge distribution in the different variable regions. Color image available online at
Discussion
The characteristics of the HIV-1 gp160 (viz. distinct biological, immunological, and pathogenic properties of different clades of HIV-1) and its role in immune evasion during horizontal transmission 22,23 has been reported previously. 24 While it is possible that vertically transmitted variants might share some of these properties, there are but few and controversial results. 25 It is reported that in MTCT, a huge volume of immune pressure is shared between the mother and the child. 26 This could insinuate adaptive cellular immune responses to the virus in the infant, as immune escape mutations in HIV-1 generated in the mother may have been transmitted to the child. 26 It has been reported that during MTCT, envelope variants resistant to neutralization by maternal antibodies are preferentially transmitted. 27 This dynamics of the immune pressure is likely to be the driving force for the enormous genetic diversity, variation in frequency and pattern of glycosylation, and length of the variable loops in the vertically transmitted viral variants, and define the fitness of the organism. In this study, we describe the genetic characteristics of the envelope of HIV-1 isolates from children recently infected with HIV-1 through the vertical route. Almost all sequences were identified to use CCR5 as the co-receptor, had unique features in the variable loops and exhibited a characteristic glycosylation pattern.
Subtype analysis using online tools RIP, jpHMM, and REGA showed that all but one of the experimental sequences belonged to subtype C and had a GPGQ motif in the V3 loop. NIRT_ENV017 alone had a GPGR motif in the V3 loop and was identified as subtype A/AG. Subtyping was further confirmed by phylogenetic analysis of full-length envelope sequences. All the experimental sequences formed a tight monophyletic cluster with subtype C sequences.
Several web-based algorithms were used to predict the co-receptor usage pattern of the isolates. The bioinformatics tools used for co-receptor prediction have become more complex, but the 11/25 rule 28 [lysine/arginine (K/R) with serine/aspartic acid (S/D) at positions 11 and 25 in the V3 loop, respectively] and the overall net electric charge of the V3 region still remain the key elements of most algorithms. In addition, it is believed that change in V3 length, even by a single mutation at any position within the V3 loop, can cause the virus to switch co-receptor usage. 29 In our study, the vast majority of the isolates (15/17) were predicted to use CCR5 as the co-receptor. In one of the two isolates that were predicted as X4 tropic, lysine was present at position 25 in the V3 region of one sequence (NIRT_ENV004).
One of the env sequences (NIRT_ENV016) had a number of stop codons scattered along the entire length of the env sequence except in the variable regions. The quality of the nucleotide sequence was carefully examined and it was observed that the stop codons were introduced as a result of G-A transitions formed as a result of APOBEC3-G-mediated hypermutation. We found from literature that such a phenomenon exists in about 38% of asymptomatic individuals and 50% of seroconverters. 30 The stop codons found in this sequence could make the virus both infection and replication incompetent. 30 Interestingly, G-A hypermutations were not seen in the variable regions. This could be the result of positive selection by immune pressure. However, the functional relevance of G-A hypermutation in the clinical consequence of disease progression remains inconclusive.
Previous reports have shown that during the course of the disease with different subtypes of the M group, 31 changes in both amino acid sequence, length and number of PNGS in the V1, V2, and V4 regions, generate HIV variants resistant to neutralizing antibodies. 22 Besides, resistance to neutralizing antibodies could possibly be also due to shielding of the underlying regions of the envelope glycoprotein from antibody recognition. 8 The length and glycosylation characteristics of the variable regions appear to play a vital role in HIV-1 transmission, co-receptor usage and viral entry, 22 disease progression, 32 and fusion efficiency. 33 Longer V1V2 loops with an increased number of PNGS are thought to be associated with increased neutralization resistance among recently transmitted HIV-1 from contemporary seroconverters. 6,25 It has been shown that deletion of the V1V2 loop can result in a more open conformation of the envelope, with better exposure of the V3 loop, and that even relatively small changes in the V1V2 stem may result in a more open envelope conformation, making the virus more susceptible to anti-V3 antibodies. 7
The third variable region of gp120, is known to be variable in subtypes B and D but relatively conserved in HIV-1 subtypes A, C, G, E, and H 22 and plays an important role in defining co-receptor usage, 34 neutralization, cellular immunity, 35 and env-mediated fusion. 22,36 The isolates examined in this study also showed a fair degree of conservation of amino acid sequence in the V3 region. The V3 region is also identified as a potent neutralizing determinant for T cell line adapted strains of HIV-1, and it is thought to be an important component of an HIV-1 vaccine. 36 Differences in infectivity fitness due to mutation at position 9 in the V3 region has been reported previously; switch to higher infectivity was identified in isolates with arginine at this position. 29 Interestingly, almost all but one of the sequences obtained from our study subjects had arginine at position 9 (Fig. 2). We speculate therefore that the isolates obtained from recently infected individuals possess higher infectivity fitness. Another important finding is the enormous genetic diversity in the V4 region. This has been previously reported to be associated with disease progression in simian immunodeficiency virus (SIV) and HIV infection. 32 Moore et al. also observed enormous variation in the V4–V5 regions and associated it with response to immune pressure. 37 Further, extensive mutation in the V4 region has also been reported to be associated with co-receptor switching from R5 to R5X4. 34 In contrast to horizontal transmission, V5 regions of vertically transmitted envelopes showed broader difference in length with insertions and deletions.
Infectivity fitness of the virus variant is also known to depend on the extent of glycosylation, and the pattern of glycosylation. 17 Glycosylation is the process of acquiring carbohydrate side-chains as a post-transcriptional modification, so as to form a tight glycan shield that can mask the epitopes from antibodies, and help to overcome host immune defences. 38 We found an increased frequency of PNGS (average being around 30) in the env sequence of vertically transmitted viral isolates. This finding is in contrast to previous reports that the gp160 of horizontally transmitted viral isolates, after adaptation, contain fewer numbers of PNGS (∼20–25). 20 The higher frequency of PNGS seen in the vertically transmitted envelope sequence could have resulted either from forward mutations in the child, or may have already occurred in the mother before transmission to the child.
In general, mutations that occur during the establishment of infection induce changes in the positioning of glycans on gp160, and alter the antigenic makeup of the viral envelope. In our study, we found deletion and addition of glycans in the variable regions. Viral mutants that lack variable loop glycosylation sites are reported to be more susceptible to neutralization. 39 In addition, the loss of PNGs can also impact the immunogenicity of noncovalently associated proteins. In particular, abrogation of glycans in the V1V2 region alters the tertiary structure of gp120 and could eventually result in altered infection fitness. Deletion of glycans in and around the V3 loop can result in switching of viral tropism and increased infectivity fitness. 17 Glycosylation of residues in the V5 region is thought to revert rapidly in almost 75% of the sequences, which indicates a fitness disadvantage of the mutant forms. We hypothesize that the increased levels of glycosylation in our isolates was possibly due to transmitted mutations from the mother to the child.
Alteration of glycosylation sites can have dramatic consequences for a virus. It can impact protein folding and affect distant parts of the protein through masking or conformational changes. Moreover, altered patterns of glycosylation in viral proteins can contribute to escape from T cell responses and influence receptor binding and phenotypic properties of viruses. Such an association was identified in children isolates with minimal increase in the frequency of NNTT glycosylation pattern (data not shown). NNTT is a common pattern among HIV sequences, where steric occlusion may prevent addition of carbohydrates to both asparagines. 3
In addition to the above properties, we also examined the net electric charge of the different variable regions, as this feature is again thought to have a role in driving HIV-1 infection. It has previously been reported that the difference in amino acid electric charge was closely associated with resistance to neutralizing antibodies. 40 During HIV-1 transmission, under immune pressure, the carboxyl and amino group of the amino acid will ionize giving the viral capsid a net electric charge 41 that enables fusion of the virus with the host cell. Changes in net electric charge and physical interactions between the variable regions can affect the structural conformation of the env during the course of disease. These changes may be involved in the virus escape mechanism.
It has been previously reported that the V2 region of the env interacts with the integrin α4β7 receptor of the host cell. For virus capture such an interaction is essential. However, induction of antibodies that block this mechanism could play an important role in preventing acquisition of HIV-1 infection. 42 In our study it was observed that the net electric charge of the V2 and V3 regions was stable. However, significant difference in electric charge was identified in the V1, V4, and V5 regions. In contrast, in horizontal transmission, stable charges have been reported in the V1 and V4 regions. 22
It is well known that the net charge of the V3 loop can determine HIV-1 tropism and co-receptor usage. 36 Based on net charge, all but one of the study sequences, were identified to use CCR5 as the co-receptor at the time of infection. Only one sample (NIRT_ENV017) had a neutral net electric charge in the V3 region, and was predicted to use CXCR4 as the co-receptor. It has been previously reported that conserved amino acid sequence length, number of PNGS, and net electric charge of the V5 region play a crucial role in determining the three-dimensional structure of the env and influence glycan packing and immune evasion. 37 In our study, we observed that vertically transmitted env sequences showed divergence of amino acids with deletion of PNGS in the V5 region (13 of the 17 env sequences did not have any PNGS in the V5 region), and a difference in the net electric charge.
Conclusion
In brief, our study revealed that majority of the proviral env sequences obtained from HIV+ infants following vertical transmission, had unique characteristic features in their variable regions and glycosylation patterns. Interestingly, total lack of PNGS was observed in the V5 region of majority of the variants that distinguishes them from horizontally transmitted HIV strains. On the other hand, a stable net electric charge was observed in the V2 and V3 regions, and is thought to be associated with their involvement in viral fusion and co-receptor tropism. In addition, APOBEC3-G mediated G-A hypermutation derived stop codon was identified for the first time in a vertically transmitted viral variant in the early stages of infection. Further, the difference in the length of the variable region, characteristic pattern of deposition of PNGs and their frequency throughout the envelope, and net electric charge of the variable loops observed in this study require phenotypic analysis to identify the unique quasispecies that is responsible for the spread of HIV in the early stages of infection through the vertical route.
Data deposition
The sequences reported in this article have been deposited in the GenBank database (accession nos. KX756600–KX756615).
KX756600—KX756614 and KX756615 correspond to sample ids, NIRT_ENV001–NIRT_ENV015 and NIRT_ENV017, respectively.
The sequence, NIRT_ENV016 was not deposited in GenBank due to presence of stop codons. The sequence is provided as Supplementary Fig. S2.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.