
Abstract
Effector memory T cell (TEM) responses display potent antiviral properties and have been linked to stringent control of simian immunodeficiency virus (SIV) replication. Since recurrent antigen stimulation drives the differentiation of CD8+ T cells toward the TEM phenotype, in this study we incorporated a persistent herpesviral vector into a heterologous prime/boost/boost vaccine approach to maximize the induction of TEM responses. This new regimen resulted in CD8+ TEM-biased responses in four rhesus macaques, three of which controlled viral replication to <1,000 viral RNA copies/ml of plasma for more than 6 months after infection with SIVmac239. Over the course of this study, we made a series of interesting observations in one of these successful controller animals. Indeed, in vivo elimination of CD8αβ+ T cells using a new CD8β-depleting antibody did not abrogate virologic control in this monkey. Only after its CD8α+ lymphocytes were depleted did SIV rebound, suggesting that CD8αα+ but not CD8αβ+ cells were controlling viral replication. By 2 weeks postinfection (PI), the only SIV sequences that could be detected in this animal harbored a small in-frame deletion in nef affecting six amino acids. Deep sequencing of the SIVmac239 challenge stock revealed no evidence of this polymorphism. However, sequencing of the rebound virus following CD8α depletion at week 38.4 PI again revealed only the six-amino acid deletion in nef. While any role for immunological pressure on the selection of this deleted variant remains uncertain, our data provide anecdotal evidence that control of SIV replication can be maintained without an intact CD8αβ+ T cell compartment.
Introduction
T
Herpesviruses establish latent infections that persist for the life of the host. 20 Similar to live-attenuated SIV vaccination, herpesviral infections result in persistent Ag stimulation, which favors the induction of effector memory T cell (TEM) responses. 21 This phenotype is associated with T cells that recirculate through extralymphoid tissues and are poised for immediate antiviral activity. 22 The persistent nature of herpesviruses and the antiviral properties of TEM prompted the generation of live recombinant (r) herpesviral vectors encoding SIV proteins. For example, a fibroblast-adapted strain of the β-herpesvirus rhesus cytomegalovirus (RhCMV) expressing SIV inserts has shown great promise in monkey trials with approximately half of RhCMV/SIV vaccinees manifesting early and profound control of viral replication after SIVmac239 infection. 23,24 Remarkably, these protected vaccinees eventually cleared SIV in vivo, implying that lentiviral infections are vulnerable to early interception by vaccine-induced TEM responses. 25 Importantly, RhCMV is not the only herpesviral vector platform available for persistent Ag delivery in nonhuman primates. In fact, a genetic system for the γ2-herpesvirus rhesus monkey rhadinovirus (RRV) has also been developed, 26 facilitating the generation of rRRV/SIV vectors. 27 In contrast to the unconventionally major histocompatibility complex (MHC)-restricted CD8+ TEM responses elicited by the aforementioned RhCMV strain, 28,29 rRRV/SIV-vaccinated macaques develop CD8+ T cells capable of recognizing classically immunodominant MHC class I-restricted SIV epitopes. 27 Following intravenous challenge with SIVmac239, rRRV/SIV vaccinees manifested significant reductions in peak and set point viremia compared to unvaccinated controls. Collectively, these data highlight the utility of herpesviral vectors for eliciting lentivirus-specific TEM responses.
Repeated Ag stimulation delivered in the form of heterologous prime/boost/boost (PBB) immunization protocols has been shown to generate robust CD8+ TEM responses in mice. 30,31 Similarly, we have recently shown that a rDNA prime followed by boosting with rYF17D and rAd5 resulted in high-frequency SIV-specific TEM responses in rhesus macaques. 32 However, these vaccine-induced CD8+ T cells underwent considerable contraction soon after the rAd5 boost, possibly due to the lack of persistent Ag stimulation afforded by the rDNA/rYF17D/rAd5 protocol. In an attempt to improve the maintenance of CD8+ TEM responses, we delivered a final rRRV/SIV boost to four rhesus macaques that had been immunized with an rDNA prime followed by a rYF17D boost. In addition to evaluating the immunogenicity and efficacy of this new rDNA/rYF17D/rRRV regimen, this pilot study resulted in an extraordinary outcome in one of the vaccinated monkeys. Using viral sequencing and selective depletions of CD8β- or CD8α-expressing lymphocytes in vivo, we investigated immunological and virologic aspects of this rare case of control of SIVmac239 replication.
Materials and Methods
Research animals
The four Indian rhesus macaques used in the vaccine pilot study described in Figure 1 were naturally infected with RRV. The two SIV-infected Indian rhesus macaques used in the experiment described in Figure 4 were part of a recent study conducted by our laboratory and were used to further characterize the effects of CD8β depletion during controlled SIV infection. All animals were housed at the Wisconsin National Primate Research Center (WNPRC) and cared for in accordance with the Weatherall Report under a protocol approved by the University of Wisconsin Graduate School Animal Care and Use Committee. Vaccinations, SIV challenges, and monoclonal antibody (mAb) infusions were performed under anesthesia, and all efforts were made to minimize potential suffering. Additional animal information, including MHC class I alleles, age, and sex, is shown in Table 1.
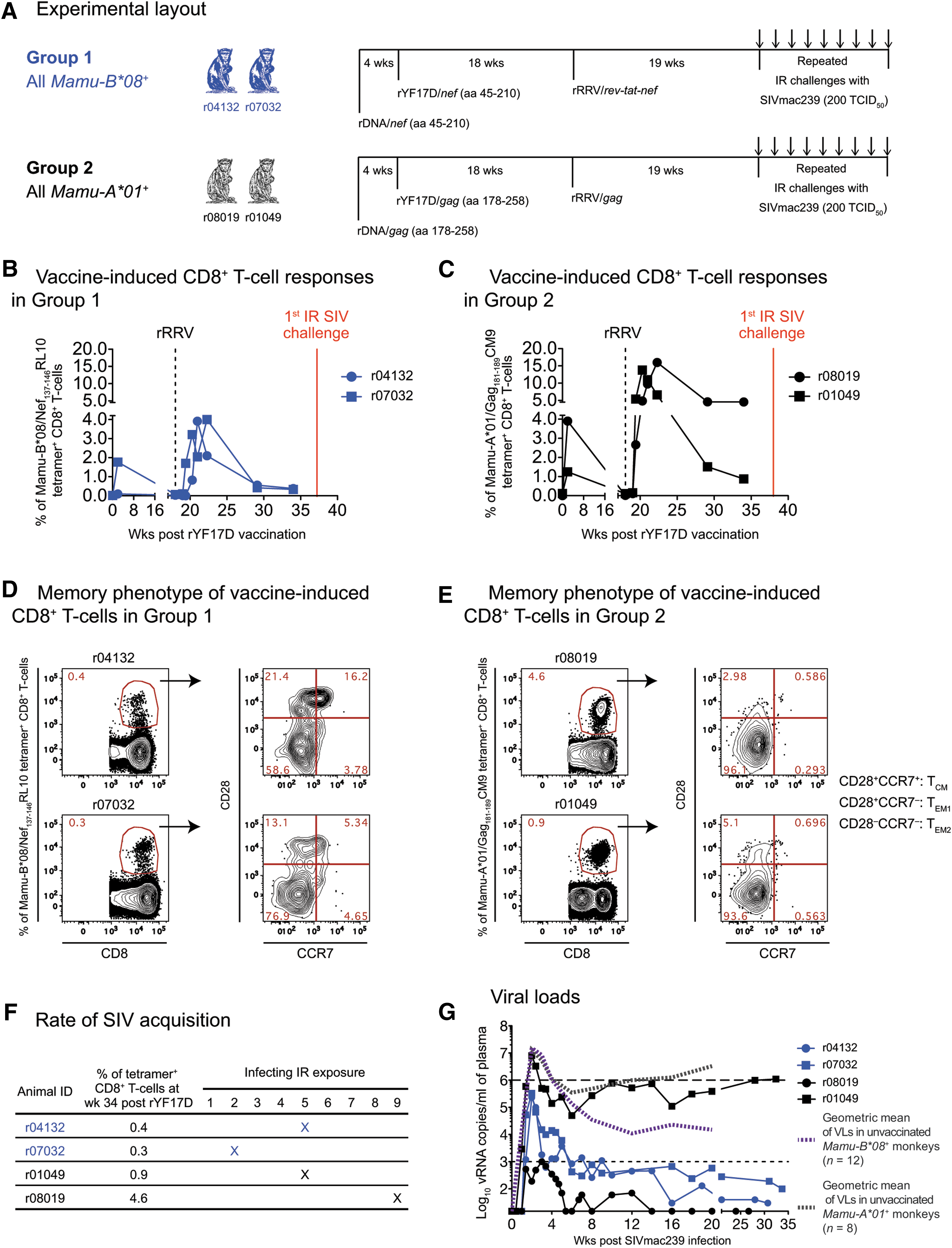
Design, immunogenicity, and efficacy of an rDNA/rYF17D/rRRV regimen encoding immunodominant classical CD8+ T cell epitopes.
Animal age at the beginning of study.
MHC, major histocompatibility complex.
Vaccinations
The macaques in Groups 1 and 2 were primed once with rDNA plasmids expressing SIVmac239 minigenes encoding Nef amino acids (aa) 45-210 (Group 1) or Gag aa 178-258 (Group 2). One milligram of each rDNA/SIV vector and 0.1 mg of an IL-12-expressing plasmid were codelivered by intramuscular electroporation. The monkeys then received 300,000 plaque-forming units of rYF17D vectors encoding the same inserts as the rDNA plasmids through the subcutaneous route. Additional information on the rDNA and rYF17D vaccinations can be found elsewhere. 33 The third and last vaccine boost consisted of a single infusion of 1.0 mL of phosphate-buffered saline (PBS) solution containing 7.0 × 108 genome copies of rRRV vectors encoding SIVmac239 inserts. Animals in Group 1 received rRRV/rev-tat-nef while those in Group 2 were vaccinated with rRRV/gag (Fig. 1A). The construction of these rRRV/SIV vaccines has been described elsewhere. 27
SIVmac239 challenges
The challenge stock utilized in this study was produced by the Virology Services Unit of the WNPRC using SIVmac239 hemi-genome plasmids obtained from the NIH AIDS Research and Reference Reagent Program. These plasmids were transfected into 293T cells, and the supernatant was propagated on mitogen-activated peripheral blood mononuclear cell (PBMC) from SIV naive rhesus macaques for several days. The titer of this stock was 90,000 50% tissue culture infective doses (TCID50)/mL. Animals in this study were subjected to the same weekly intrarectal (IR) SIVmac239 challenge regimen described recently. 32 The dose of each exposure was 200 TCID50, which corresponded to 4.8 × 105 viral RNA (vRNA) copies. Plasma viral loads (VLs) were assessed 3 and 7 days after each exposure. Once an animal had a positive VL, it was no longer challenged.
In vivo CD8 depletions
CD8β-expressing lymphocytes were transiently depleted using the anti-CD8β mAb CD8β255R1. The parental mouse mAb was raised against rhesus CD8β chain and engineered into a recombinant rhesus IgG1 by complementarity determining region (CDR) grafting. CD8α+ cells were depleted using a CDR-grafted rhesus IgG1, MT807R1, derived from a mouse anti-human CD8α mAb. 34 All animals were administered a single intravenous injection of 50 mg/kg of body weight. Both mAbs were provided by the NIH Nonhuman Primate Reagent Resource (Boston, MA).
MHC class I tetramer staining and immunophenotyping during lymphocyte depletions
We monitored the ontogeny of vaccine-induced SIV-specific CD8+ T cell responses in Groups 1 and 2 using fluorochrome-labeled MHC class I tetramers, as described recently. 35 In sum, PBMCs were isolated from blood drawn at the WNPRC on the previous day and then shipped to the University of Miami overnight. We used allophycocyanin-conjugated Mamu-B*08/Nef137-146RL10 (NIH Tetramer Core Facility) and Mamu-A*01/Gag181-189CM9 (MBL International) tetramers to monitor vaccine-induced SIV-specific CD8+ T cell responses in the Group 1 and Group 2 macaques, respectively. Up to 800,000 PBMCs were incubated with titrated amounts of each tetramer at 37°C for 1 h and then stained with fluorochrome-labeled antibodies directed against the surface molecules CD3 (clone SP34-2), CD8α (clone RPA-T8), CD28 (clone 28.2), CCR7 (clone 150503), CD14 (clone M5E2), CD16 (clone 3G8), and CD20 (clone 2H7). Amine-reactive dye (ARD; LIVE/DEAD Fixable Aqua Dead Cell Stain; Life Technologies) was also added to this mAb cocktail. After a 25-min incubation at room temperature (RT), the cells were washed with Wash Buffer (Dulbecco's PBS with 0.1% bovine serum albumin and 0.45 g/L NaN3) and then fixed with PBS containing 2% of paraformaldehyde. The configuration of the Special Order Product BD LSR II cytometer used to acquire the samples and the gating strategy used to analyze the data have been detailed elsewhere. 32 In sum, we used FlowJo 9.6 to determine the percentages of live CD14−CD16−CD20−CD3+CD8+tetramer+ lymphocytes displayed on Figure 1B, C and to delineate memory subsets within tetramer+ populations (Fig. 1D, E).
To monitor the frequencies of lymphocyte subsets during the CD8β and CD8α depletions, our surface staining master mix included the same clones of anti-CD3, anti-CD14, anti-CD16, and anti-CD20 antibodies and the same ARD reagent mentioned above. In addition, this cocktail included antibodies against the CD8α chain (clone SK1 for the CD8β depletion phase or clone DK25 for the CD8α depletion phase) and the CD8αβ heterodimer (clone 2ST8-5H7). This latter clone recognizes a conformational epitope consisting of domains from both the CD8α and CD8β chains. 36 Of note, the CD8 glycoprotein can be expressed as two isoforms: CD8αβ heterodimers or CD8αα homodimers. 37 The anti-CD8α mAb MT807R1 that was used to deplete CD8α+ lymphocytes in vivo is known to cross-block other anti-CD8 antibodies used for immunophenotyping. However, the anti-CD8α clone DK25 can still resolve CD8α+ cells in the presence of MT807R1, although with reduced fluorescence intensity. 34 In quality assessment tests performed in our laboratory, we found that preincubation of PBMC with 50 μg/mL of the anti-CD8β mAb CD8β255R1 did not prevent fluorochrome-labeled 2ST8-5H7 antibodies from detecting CD8αβ+ T cells, although there was a slight reduction in fluorescence intensity (data not shown). In contrast, preincubation of PBMC with 50 μg/mL of MT807R1 completely blocked staining with 2ST8-5H7. During the CD8β depletion phase, CD3+ T lymphocytes that were positive for CD8α but negative for CD8αβ were considered to be CD8αα+. To obtain relative frequencies of lymphocyte subsets, we first excluded monocytes, B cells, and dead cells in the “dump gate” (CD14+CD20+ARD+) and then determined the percentages of CD8αβ+ T cells (CD3+CD8αα−CD8αβ+), CD8αα+ T cells (CD3+CD8αα+CD8αβ−), CD4+ T cells (considered as CD3+CD8αα−CD8αβ−), and NK cells (CD3−CD8αα+CD16+) within the lymphocyte gate. Absolute counts of these subsets were then calculated by multiplying their respective frequencies by the absolute number of white blood cells obtained from matching complete blood counts.
Intracellular cytokine staining assay
The intracellular cytokine staining (ICS) assay was performed as described recently. 32 In sum, cells were stimulated with SIV peptides at a final concentration of 1.0 μM in the presence of costimulatory mAbs against CD28 (clone L293; BD Biosciences) and CD49d (clone 9F10; BD Pharmingen) for 9 h at 37°C in a 5.0% CO2 incubator. To inhibit protein transport, Brefeldin A (BioLegend, Inc.) and GolgiStop (BD Biosciences) were added to all tubes 1 h into the incubation period. We used the same steps outlined above to stain molecules on the surface of cells and to fix them with 2% of paraformaldehyde. In addition to the same mAbs against CD14, CD16, and CD20 and the ARD reagent described above, the surface staining master mix also included mAbs against CD4 (clone OKT4; BioLegend, Inc.) and CD8 (clone RPA-T8; BioLegend, Inc.). Cells were permeabilized by resuspending them in “Perm Buffer” [1 × BD FACS lysing solution 2 (Becton Dickinson) and 0.05% Tween 20 (Sigma-Aldrich)] for 10 min and subsequently washed with Wash Buffer. Cells were then incubated with mAbs against IFN-γ (clone 4S.B3; BioLegend, Inc.), TNF-α (clone Mab11; BD Biosciences), and CD3 (same one mentioned above) for 1 h in the dark at RT, washed, and subsequently stored at 4°C until acquisition.
Anti-immunoglobulin enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) plates (Nunc-Immuno) were coated overnight at 4°C with either MT807R1 or CD8β255R1. All subsequent steps and incubations were performed at RT. On the following day, plates were washed five times with Wash Buffer (PBS containing 0.05% of Tween 20) and then blocked with SuperBlock buffer (Thermo Scientific) for 15 min. Plasma from r04132 and r08019 collected 8 days before (predepletion) or 22 days after (postdepletion) the anti-CD8β mAb infusion was used in this assay. Plasma from untreated or anti-CD8α-treated monkeys was used as negative and positive controls, respectively. After diluting plasma samples 10-fold in PBS containing 2% of bovine serum albumin, these aliquots were serially diluted 1:4 until a dilution factor of 10,240 was achieved. One hundred microliters of these serially diluted plasma aliquots were added to plates, followed by a 1-h incubation. Subsequently, plates were washed five times, and 100 μL of a 100-fold dilution of anti-immunoglobulin (Ig) human λ light chain conjugated to biotin (Miltenyi Biotec) was added to each well and incubated for 1 h. Plates were then washed five times before 100 μL of a 10,000-fold dilution of horseradish peroxidase-conjugated Streptavidin (Invitrogen) was added to each well. After a 1-h incubation, plates were washed five times. One hundred microliters of TMB (Calbiochem) was added to the plates for ∼3 min and then the substrate reaction was stopped with H2SO4 Stop Solution (Southern Biotech). The optical density (OD) of each well was determined on an ELISA reader using a 450 nm filter. End point titers of anti-Ig antibodies were considered as the highest dilutions where the OD of the postdepletion sample was >2-fold higher than its predepletion counterpart.
SIV VL measurements
VLs were measured using 0.5 mL of EDTA-anticoagulated plasma based on a modification of a previously published protocol. 38 Total RNA was extracted from plasma samples using QIAgen DSP virus/pathogen Midi kits, on a QIAsymphonyXP laboratory automation instrument platform. Six replicate two-step reverse transcription-polymerase chain reaction (RT-PCR) were performed per sample using a random primed reverse transcription reaction, followed by 45 cycles of polymerase chain reaction (PCR) using the following primers and probe: forward primer: SGAG21: 5′-GTCTGCGTCAT(dP)TGGTGCATTC-3′; reverse primer SGAG22: 5′-CACTAG(dK)TGTCTCTGCACTAT(dP)TGTTTTG-3′; probe: PSGAG23: 5′-FAM-CTTC(dP)TCAGT(dK)TGTTTCACTTTCTCTTCTGCG-BHQ1-3′. The limit of reliable quantitation on an input volume of 0.5 mL of plasma was 15 vRNA copies/ml.
Whole-genome deep sequencing of SIVmac239
vRNA in plasma was isolated using the QIAamp vRNA Mini Kit (QIAGEN), according to the manufacturer's protocol, and eluted in 60 μl of AVE buffer. The eluate was then aliquoted and stored at −80°C for future use. vRNA was reverse transcribed and amplified using the SuperScript III One-Step RT-PCR system with High Fidelity Platinum Taq polymerase (Invitrogen). PBMCs from rhesus macaque were washed twice with PBS, and DNA was then extracted from the cells using the QIAamp DNA Blood Mini Kit (QIAGEN) with PCR amplifications performed as described below with the exception of the first RT step. For the r08019 week 38.4 postinfection (PI) sample, a near full-length genome of SIV was amplified in four segments using the following primers: Amp1-F (5′-AGGAACCAACCACGACGGAG-3′) and Amp1-R (5′-AAAGGGATTGGCACTGGTGCGAGG-3′); Amp2-F (5′-TGCTGACGGCTTGTCAAGGAGTAGG-3′) and Amp2-R (5′-ACCCTGTCATGTTCCAGGTCTGTCC-3′); Amp3-F (5′-ATGGTGGGCAGGGATAGAGC-3′) and Amp3-R (5′-CCATGCCTGCTTTGGCCTAT-3′); and Amp4-F (5′-TGCACAGGCTTGGAACAAGA-3′) and Amp4-R (5′-ACATCCCCTTGTGGAAAGTC-3′). The RT-PCR conditions were as follows for Amp 1 and 2: 50°C for 30 min; 94°C for 2 min; 40 cycles of 94°C for 15 s, 55°C for 30 s, and 68°C for 3 min; and 68°C for 5 min and for Amp 3 and 4: 45°C for 120 min, 94°C for 15 s; 40 cycles of 94°C for 15 s, 50°C for 30 s, and 68°C for 6 min; and 68°C for 6 min. In the case of PBMC from r08019 collected at week 2 PI, only one amplicon was successfully amplified and sequenced, despite multiple attempts using different primer sets and conditions. A 2.4-kb fragment spanning part of env and nef (positions 7,565–9,979) amplified using forward primer (5′-CAGTCACCATTATGTCTGGATTG-3′) and reverse primer (5′-GAATACAGAGCGAAATGCAGTG-3′) under the conditions detailed above for amp 1 and 2. A smaller amplicon-based 454 sequencing approach was used to analyze a 400-bp region in Nef as previously described. 39 Whole genome deep sequencing was also performed on the SIVmac239 challenge as previously described. 40
Amplicons were visualized on a 1.0% agarose gel and purified using the PureLink Quick Gel Extraction Kit (Invitrogen). RT-PCR products were quantified using a Promega QuantiFluor-ST fluorometer (Promega) and analyzed for quality using an Agilent 2100 Bioanalyzer with high sensitivity DNA chips. PCR amplicons were fragmented and bar coded using Nextera XT DNA Library Prep Kit, as per manufacturer's protocol. Samples were pooled and sequenced on an Illumina MiSeq platform, using a 2 × 250-bp V2 Reagent Kit. Paired-end reads obtained from Illumina MiSeq were assembled into a SIV consensus sequence using the VICUNA de novo assembler software and finished with V-FAT v1.0. 41 Reads were mapped back to this consensus using Mosaik v2.1.73 and intrahost variants called by V-Phaser v2.0. 42,43 All reads have been deposited to the NCBI Sequence Read Archive under accession number SRP016012.
Results
Four rhesus macaques were equally divided between two groups based on their MHC class I alleles: monkeys in Group 1 were Mamu-B*08 + and those in Group 2 were Mamu-A*01 + (Fig. 1A). We chose these MHC class I alleles because their gene products present immunodominant SIV epitopes. Specifically, Mamu-B*08 binds to Nef137-146RL10, while Mamu-A*01 restricts Gag181-189CM9. 44,45 Of note, expression of Mamu-B*08 has been linked to elite control of SIVmac239 infection. 46
To achieve the continual Ag stimulation that favors induction of TEM responses, 21,47 we combined the repetitive boosting provided by heterologous PBB regimens and the persistent Ag exposure afforded by herpesviral infections into a new immunization protocol. This protocol consisted of successive vaccinations with electroporated rDNA, rYF17D, and rRRV vectors encoding SIVmac239 inserts (Fig. 1A). The vectors used in Group 1 and Group 2 delivered the Nef137-146RL10 and Gag181-189CM9 epitopes, respectively, to match the expressed MHC class I alleles (Fig. 1A). To monitor the ontogeny of vaccine-induced CD8+ T cell responses against each epitope, we performed longitudinal fluorochrome-labeled MHC class I tetramer stainings in PBMC from the Group 1 and Group 2 animals. The two Mamu-B*08 + vaccinees developed low frequency responses against Nef137-146RL10, with percentages of tetramer+CD8+ T cells peaking at 4% and then decreasing to <1% of CD8+ T cells by week 34 post rYF17D (Fig. 1B). Conversely, both Mamu-A*01 + animals in Group 2 mounted robust Gag181-189CM9-specific CD8+ T cells after the rRRV boost (14% and 16% at peak; Fig. 1C). While the magnitude of these tetramer+CD8+ T cells eventually declined to <1% in r01049, the other Group 2 vaccinee (r08019) maintained its Gag181-189CM9-specific response at 4.6% after resolution of the peak response (Fig. 1C). It is not entirely clear why the contraction of vaccine-induced SIV-specific CD8+ T cells was accentuated in both Group 1 animals and in r01049 in the weeks following the rRRV boost. Given that captive rhesus monkeys can be naturally infected with RRV, 48,49 preexisting immunity to this herpesvirus may have limited the “take” of the live rRRV vectors utilized in this study. Indeed, all four monkeys in Groups 1 and 2 were seropositive for RRV at the time of the rRRV vaccination (data not shown).
We also carried out a phenotypic analysis of vaccine-induced SIV-specific CD8+ T cells at week 34 post rYF17D and found a predominance of the fully differentiated TEM2 (CD28−CCR7−) phenotype (Fig. 1D, E), indicative of persistent Ag stimulation. Transitional memory (TEM1; CD28+CCR7−) and central memory (TCM; CD28+CCR7+) tetramer+CD8+ T cells were also detected, especially in the Group 1 vaccinee r04132 (Fig. 1D). In sum, while 3/4 vaccinees had low frequencies of SIV-specific CD8+ T cells at the time of SIV challenge, r08019 was clearly an outlier since >4% of its peripheral CD8+ T cell compartment was specific for a single SIV epitope and 96% of these exhibited the TEM2 phenotype.
At week 19 post rRRV boost, we began challenging all animals intrarectally with 200 TCID50 of an in vivo-titrated SIVmac239 stock. Of note, out of the 32 SIV naive rhesus macaques that have been subjected to this IR challenge regimen (same viral stock and dose) as part of ongoing experiments conducted by our group, 78% became infected by the sixth exposure (Martins et al., unpublished observations). Remarkably, after becoming infected after the ninth IR challenge (Fig. 1F), the Group 2 vaccinee r08019 experienced a peak VL of only 1,000 vRNA copies/ml of plasma and subsequently controlled viremia to <15 vRNA copies/ml by week 5 PI (Fig. 1G). In contrast, the other Group 2 animal (r01049) became infected after the fifth challenge and failed to suppress viremia (Fig. 1F, G). Encouragingly, both Group 1 vaccinees had relatively low peak VLs and controlled chronic phase VLs to <1,000 vRNA copies/ml (Fig. 1G). As a reference, we compared VLs in Groups 1 and 2 with those from MHC class I-matched macaques that were rectally infected with SIVmac239 as part of previous and ongoing studies conducted by our laboratory (Martins et al., unpublished observations). 32,33,39 These historical controls included both unvaccinated and vaccinated monkeys (Fig. 2). Overall, the two Group 1 vaccinees experienced lower peak and set point VLs than the majority of their Mamu-B*08+ historical counterparts while r08019 exhibited the lowest levels of viremia among our cohort of Mamu-A*01+ historical controls (Fig. 2). Thus, a rDNA/rYF17D/rRRV regimen encoding immunodominant SIV epitopes resulted in stringent control of SIVmac239 replication in 3/4 vaccinees.
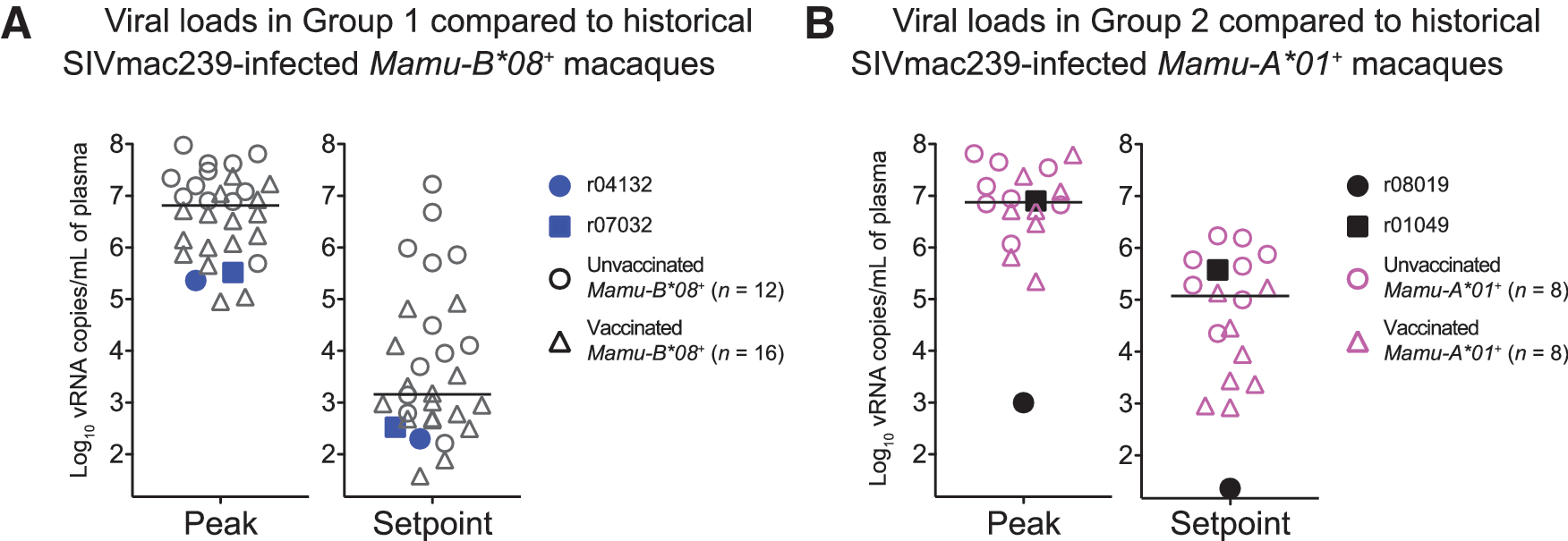
Peak and set point VLs in Groups 1 and 2 compared to historical SIVmac239-infected macaques.
Given r08019's remarkable outcome, we then explored several potential mechanisms for the impressive virologic control observed in this animal. First, we postulated that this animal's robust vaccine-induced SIV-specific CD8+ T cell response was responsible for containing viral replication. Based on this assumption and on our previous experimental data, 2 we expected r08019 to transiently lose control of SIV replication following infusion of a CD8-depleting mAb. Of interest, the CD8 glycoprotein can be expressed as two isoforms: either as a heterodimer consisting of the CD8α and CD8β chains (CD8αβ) or as a CD8αα homodimer. 37 While CD8αβ marks thymus-selected MHC class I-restricted T cells, 37,50 CD8αα is found primarily on mucosal T cells and, in the case of rhesus macaques, NK cells as well. 51,52 Historically, in vivo depletion of CD8+ lymphocytes in SIV-infected rhesus macaques has been accomplished by the infusion of antibodies specific for the CD8α chain. 5,53,54 An important caveat of this approach is the simultaneous elimination of CD8αβ+ T cells and NK cells, which makes it difficult to distinguish the contribution of each lymphocyte subset to control of SIV infection. To avoid this limitation, we used a newly available rhesus recombinant CDR-grafted mAb (CD8β255R1) specific for the CD8β chain to selectively ablate CD8αβ+ T cells in r08019 and in both Group 1 macaques (Fig. 3A). Importantly, quality assessment tests performed before the CD8β depletion confirmed that CD8β255R1 did not block the fluorochrome-labeled anti-CD8 antibodies utilized in our immunophenotyping assays (see Materials and Methods section). A single infusion of 50 mg/kg of CD8β255R1 readily depleted the majority of peripheral CD8αβ+ T cells in all three animals without eliminating NK cells (Fig. 3B, C). In fact, NK cell numbers steadily increased following the CD8β255R1 infusion and peaked at week 9 post CD8β depletion (Fig. 3C). Absolute counts of CD8αα+ T cells also rose during this period, especially in r07032 (Fig. 3D). CD4+ T lymphocyte counts went up as well (Fig. 3E), possibly due to the homeostatic expansion of memory CD4+ T cells that occurs in response to CD8 depletion in vivo. 55 Similar to previous CD8α depletion studies in SIV-infected elite controller macaques, 2 elimination of CD8αβ+ T cells in the Group 1 animals resulted in a transient increase in viremia (Fig. 3F). While r04132 promptly reasserted control of viral replication, VLs in r07032 remained higher than their baseline levels for several weeks after the CD8β depletion (Fig. 3F). Interestingly, the rise in absolute counts of CD8αα+ T cells and NK cells coincided with decreases in viremia in the Mamu-B*08 + Group 1 macaques (Fig. 3C, D, F), implying that these CD8αα-expressing lymphocytes contributed to restoring virologic control in those animals. Strikingly, except for borderline VLs (up to 30 vRNA copies/ml) shortly after the CD8β depletion, r08019 did not lose control of SIV replication and had undetectable viremia by week 4.4 post CD8β depletion (Fig. 3F). Small increases in SIV-producing cells in lymph nodes were also observed in r08019 following depletion of its CD8αβ+ T lymphocytes, as determined by in situ hybridization performed on biopsies collected before and after the mAb infusion (data not shown). This strange result suggested that an intact CD8αβ+ T cell compartment was not necessary for suppressing SIVmac239 replication in r08019.
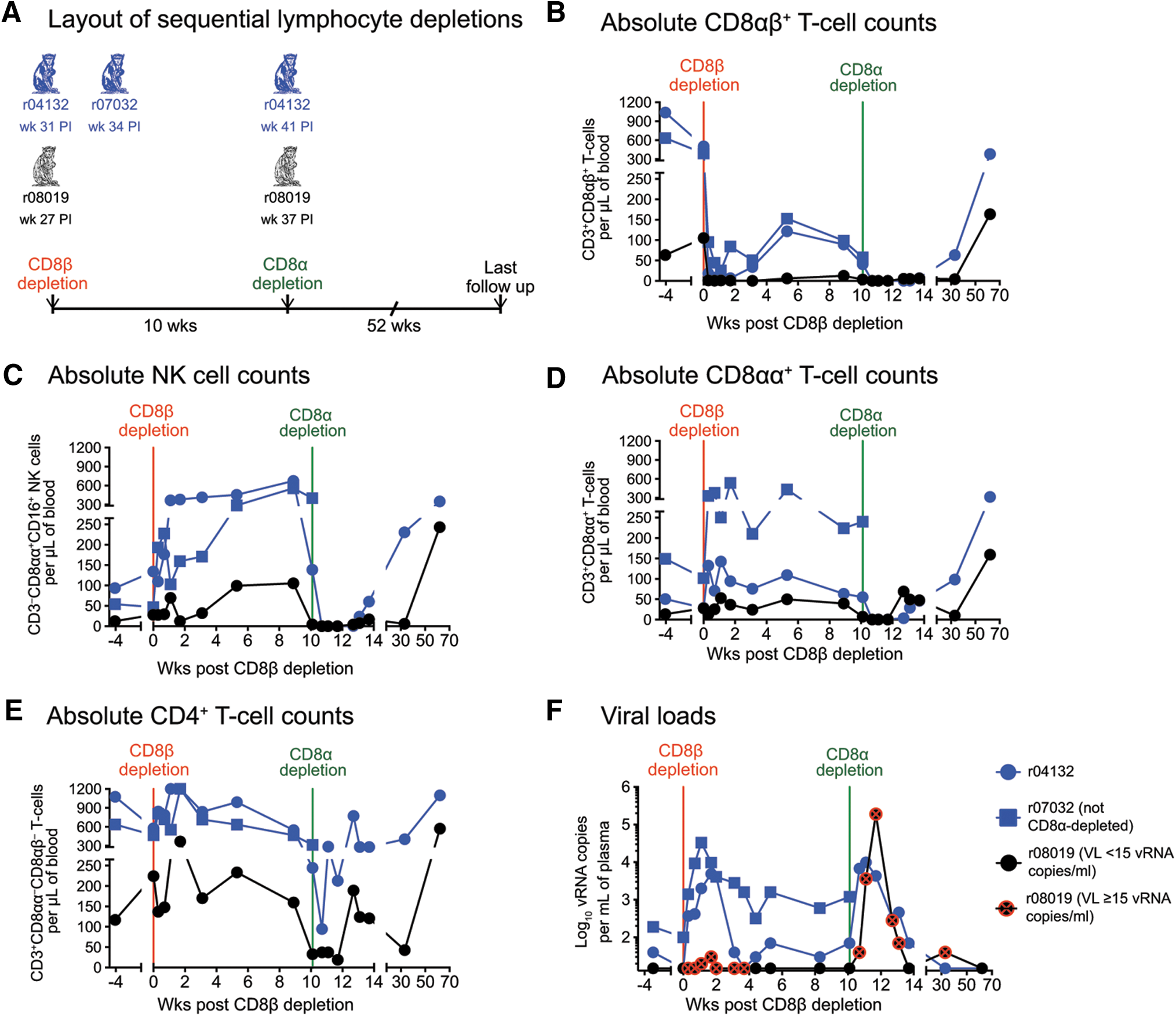
Outcome of sequential in vivo lymphocyte depletions.
To determine if the ability of r08019 to maintain control of viral replication in the context of CD8αβ+ T cell deficiency could be manifested by other macaques with controlled SIV viremia, we subjected two additional animals to the same CD8β depletion regimen described above (Fig. 4). Macaques r09089 and r09037 (Group 3) expressed Mamu-B*08+ and both controlled SIVmac239 replication as part of a recent experiment conducted by our laboratory (Table 1). 32 As described above, a single infusion of the CD8β255R1 mAb resulted in near complete elimination of peripheral CD8αβ+ T cells and a rise in NK cell counts in both r09089 and r09037, but especially in the former animal (Fig. 4A, B). Fluctuations in absolute counts of CD4+ T cells and a modest increase in CD8αα+ T cell numbers also ensued from the anti-CD8β mAb treatment (Fig. 4D, E). In contrast to r08019, but similar to the Group 1 macaques, both Group 3 macaques experienced a surge in viremia after depletion of their CD8αβ+ T cells (Fig. 4F). Although r09037 transiently regained control of viral replication at week 4 posttreatment, SIV rebounded again shortly thereafter and VLs eventually leveled off at 100–1,000 vRNA copies/ml (Fig. 4F). Monkey r09089 never reasserted full control of viral replication and its VLs ultimately plateaued in the same range as that of r09037 (Fig. 4F). These results indicated that the lack of SIV rebound manifested by r08019 in the context of CD8αβ+ T cell deficiency was not a general feature of controlled SIV infection.
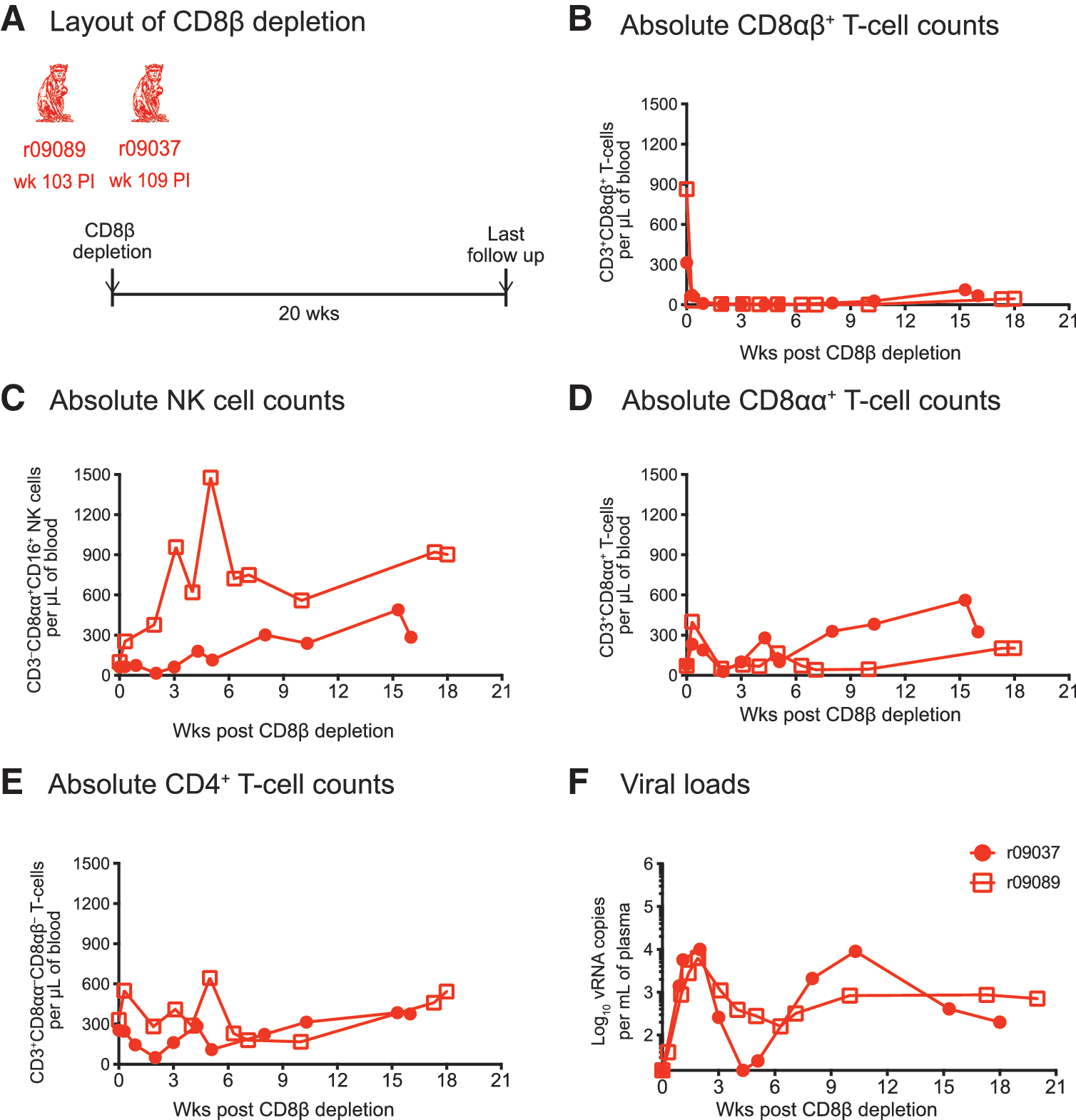
Outcome of CD8β depletion in two additional SIV-infected macaques manifesting control of chronic phase viremia.
Next, we set out to explore whether CD8αα-expressing cells prevented SIV rebound in r08019 when its CD8αβ+ T cells were eliminated. To address this possibility, we planned to treat r08019 and r04132 with the mouse/rhesus CDR-grafted anti-CD8α mAb MT807R1 at week 10 post CD8β depletion. We decided not to subject r07032 to this procedure since this animal had experienced adverse events during the CD8β255R1 infusion. A potential risk of this experiment was the development of anaphylactic reactions triggered by anti-CD8β255R1 antibodies reacting against the MT807R1 Ig molecule. To estimate the levels of these cross-reactive anti-Ig responses, we coated ELISA plates with either CD8β255R1 or MT807R1 and screened pre- and post-CD8β depletion plasma for the presence of anti-Ig antibodies. The goal of this assay was to determine the highest plasma dilution where the OD value of the postdepletion sample was >2-fold higher than the corresponding predepletion sample (i.e., the end point titer). As expected, both r04132 and r08019 developed anti-CD8β255R1 antibodies after the CD8β depletion, with end point titers of 2,560 and 160, respectively (Fig. 5). However, we could not determine the end point titer of cross-reactive anti-MT807R1 antibodies in these monkeys since their pre- and post-CD8β depletion plasma samples yielded largely equivalent OD values, even at the lowest dilution (1:10) tested (Fig. 5). Since these data suggested that r08019 and r04132 had little or no anti-MT807R1 antibodies, we decided to proceed with the CD8α depletion.
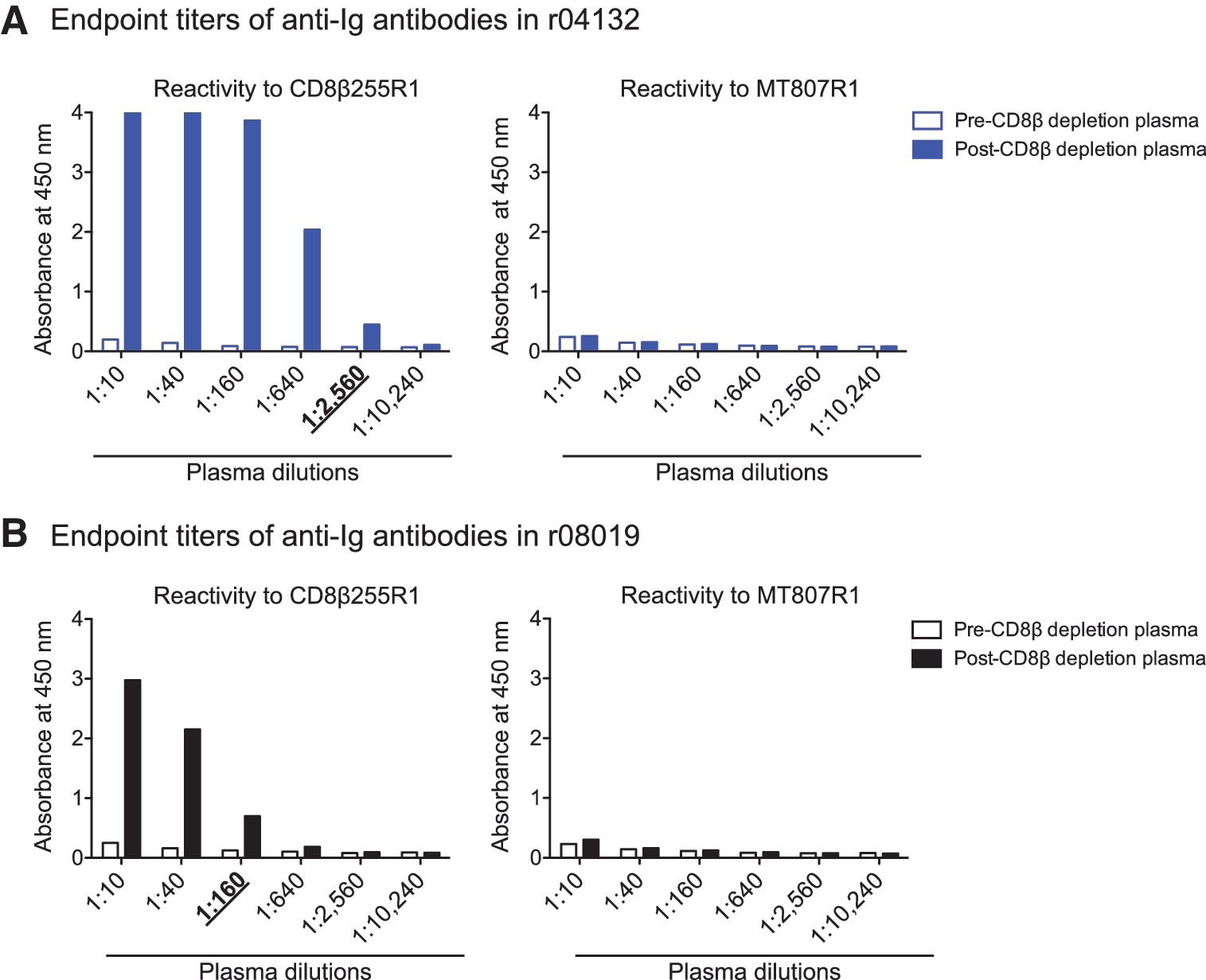
End point titers of anti-Ig antibodies in r04132 and r08019. Pre- and post-CD8β depletion plasma from r04132
Even though we could not detect anti-MT807R1 antibodies in r08019, this animal experienced decreased oxygen saturation, abnormal heart rates, and vomiting during the MT807R1 infusion. Monkey r08019 recovered after receiving supportive care, but ended up being treated with half of the planned dose of MT807R1. Conversely, r04132 did not experience any adverse reactions and received the full dose of MT807R1. Although we administered only half of the desired amount of the anti-CD8α mAb to r08019, this dose was sufficient to transiently eliminate peripheral CD8α-expressing (CD8αα+ and CD8αβ+) T lymphocytes and NK cells in this animal (Fig. 3B–D). Strikingly, SIV swiftly rebounded in r08019, peaking at 190,000 vRNA copies/ml on day 11 post CD8α depletion (week 38.4 PI; Fig. 3F). Macaque r04132 also experienced a brief rise in viremia that peaked at 9,900 vRNA copies/ml (Fig. 3F). Both animals regained control of viral replication in the ensuing weeks, concomitant with increases in absolute numbers of peripheral CD8αα+ T cells and NK cells (Fig. 3C, D, F). CD4+ T lymphocytes also expanded during this period, likely as a result of the CD8α depletion (Fig. 3E). These data suggested that effector lymphocytes expressing CD8αα homodimers, but not CD8αβ heterodimers, were responsible for maintaining virologic control in r08019.
Infection with replication-impaired lentiviruses can also lead to controlled viremia, as evidenced by the detection of defective HIV-1 variants in long-term nonprogressor patients. 56 –61 To address this possibility, we set out to determine if the virus that infected r08019 harbored any unusual insertions, deletions, or mutations. We first searched for genetic variation in proviral sequences amplified from cryopreserved PBMC collected at week 2 PI (Fig. 6A). All proviral sequences identified by this analysis contained two amino acid substitutions in Env (C432W and I863M; data not shown) and one in Nef (Y39C; Fig. 6B). In addition, an 18-bp in-frame deletion in nef corresponding to aa 139HRILDI144 was present in 100% of the sequence reads (Fig. 6B, C). Interestingly, a similar 12-bp nef deletion affecting aa 143DIYL146 is found in the SIVmacC8 clone (Fig. 6B), which has been widely used as a live-attenuated SIV vaccine. 62 –66 The highly conserved core region of Nef altered by this deletion is functionally constrained, as evidenced by the instability and functionally impaired activity of the SIVmacC8 Nef protein. 67 Moreover, SIVmac239 Nef lacking the same four amino acids that are absent from the SIVmacC8 Nef protein also exhibits the same dysfunctional phenotype. 67 Based on the similarity between the SIVmacC8 nef deletion and the deletion found in the virus discovered in r08019, we concluded that this animal harbored an attenuated SIV variant as early as week 2 PI.
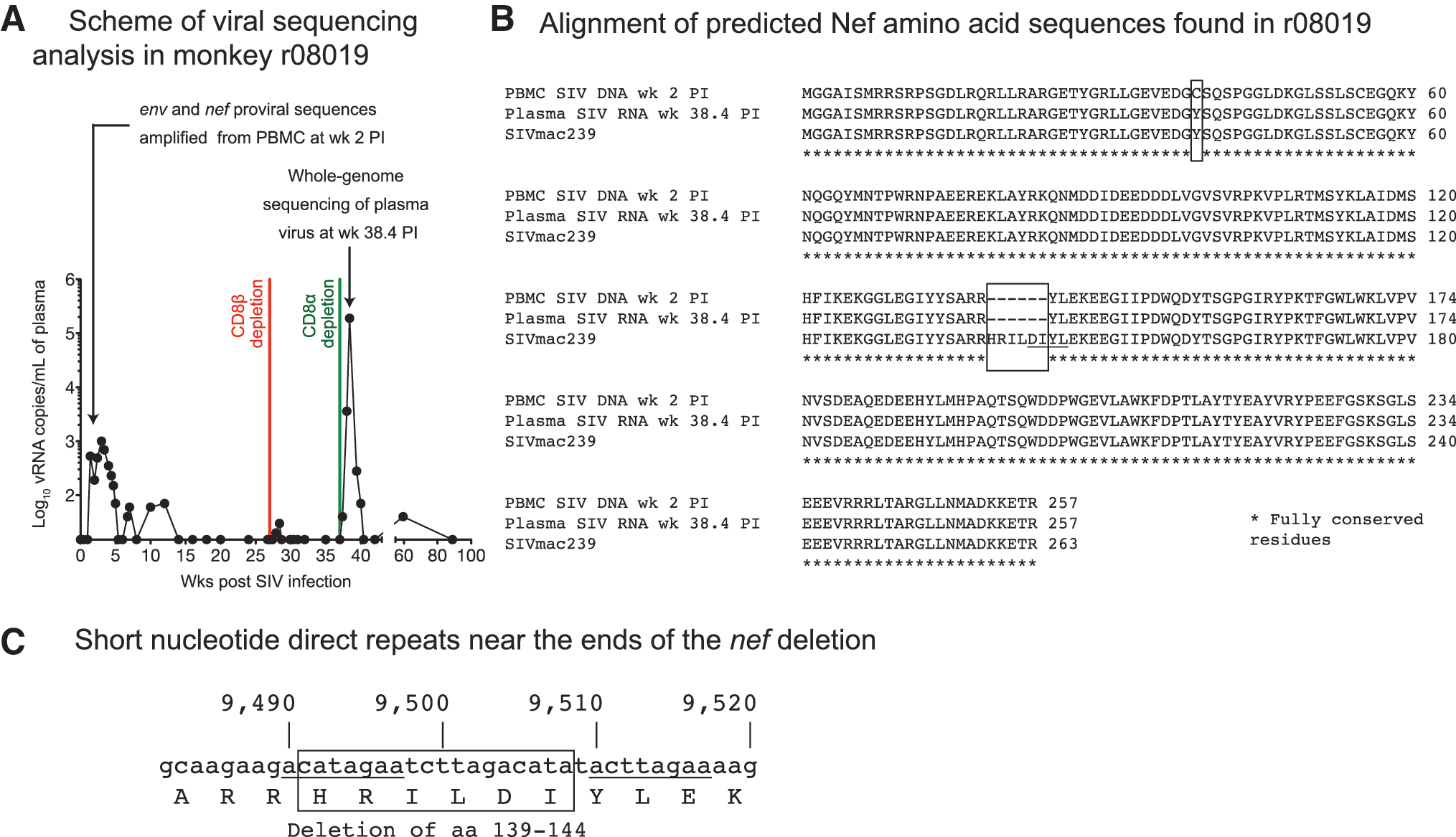
Viral sequencing analysis in r08019.
Curiously, the six amino acids affected by this deletion are contained within the Mamu-B*08-restricted Nef137-146RL10 epitope. Despite this coincidence, r08019 tested negative for Mamu-B*08 in two independent MHC typing assays. Moreover, r08019 did not have detectable Nef-specific T cell responses in an ICS assay performed at week 3.4 PI (Fig. 7), consistent with a mechanism for the emergence of the nef deletion that was independent from viral escape driven by T cell pressure.
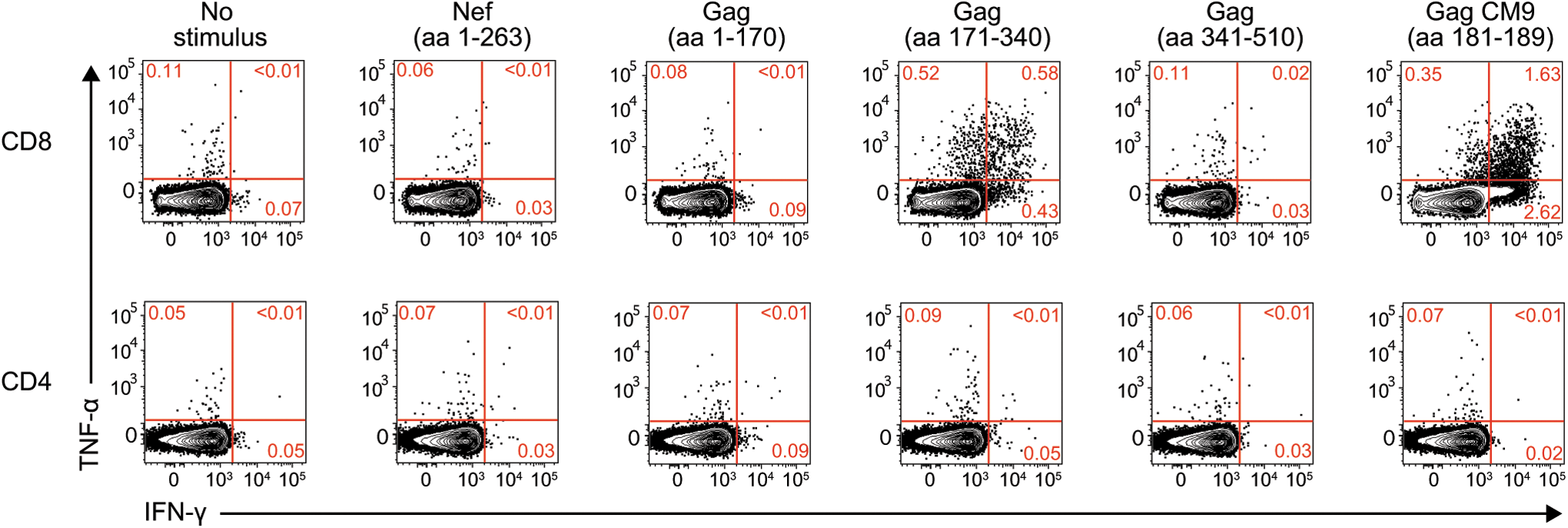
Postinfection analysis of SIV-specific T cell responses in r08019. We carried out an intracellular cytokine staining assay in PBMC from r08019 at week 3.4 PI. The stimuli consisted of pools of peptides spanning the entire Nef protein or three segments of the Gag polyprotein. A peptide corresponding to the Mamu-A*01-restricted Gag181–189CM9 epitope was included as well. Reactivity to each stimulus is shown based on the production of IFN-γ and/or TNF-α. CD8+ and CD4+ T lymphocytes are shown in the top and bottom rows, respectively.
To rule out the possibility that the nef deletion detected in r08019 was an experimental artifact, we sequenced the virus that rebounded following the CD8α depletion. Strikingly, 100% of circulating viral quasispecies contained the same deletion affecting aa 139HRILDI144 at week 38.4 PI, the peak of rebound viremia (Fig. 6A, B). These viruses also exhibited signs of CD8+ T cell-driven sequence evolution, as shown by escape mutations in the Mamu-A*01-restricted Tat28-35SL8 and Env726-735ST10 epitopes (data not shown). 68 However, the Gag181-189CM9 epitope was intact at this time point (data not shown). Of note, the Env (C432W and I863M) and Nef (Y39C) substitutions detected in cell-associated DNA at week 2 PI were absent from plasma vRNA, indicating possible reversions (data not shown; Fig. 6B). These data confirmed our previous sequencing results and showed that the nef deletion was stably maintained in the course of r08019's infection.
Finally, we investigated the origin of the nef deletion detected in r08019. Since this polymorphism was already present at week 2 PI, we explored whether the challenge inoculum contained nef-deleted SIV variants that could have initiated infection in r08019. To do that, we performed whole genome sequencing on our SIVmac239 challenge stock, which was produced by propagating the supernatant from transfected 293T cells on PBMC from SIV naive rhesus macaques over several days. This analysis revealed a relatively homogeneous virus population, with low levels of viral diversity detected across the genome (Fig. 8). Importantly, there was no evidence of the nef deletion in the stock, although the aforementioned Env (C432W and I863M) and Nef (Y39C) amino acid substitutions were present at extremely low frequencies (<0.05%). Of note, the threshold frequency for detecting mutant species in this analysis was 0.05%, considering that each sequencing reaction contained ∼2,000 copies of viral template. These results implied that either the nef deletion arose in vivo shortly after r08019 became infected or that a rare SIV variant bearing this polymorphism was present in the challenge inoculum, which went undetected by our sequencing approach, and somehow managed to infect r08019.
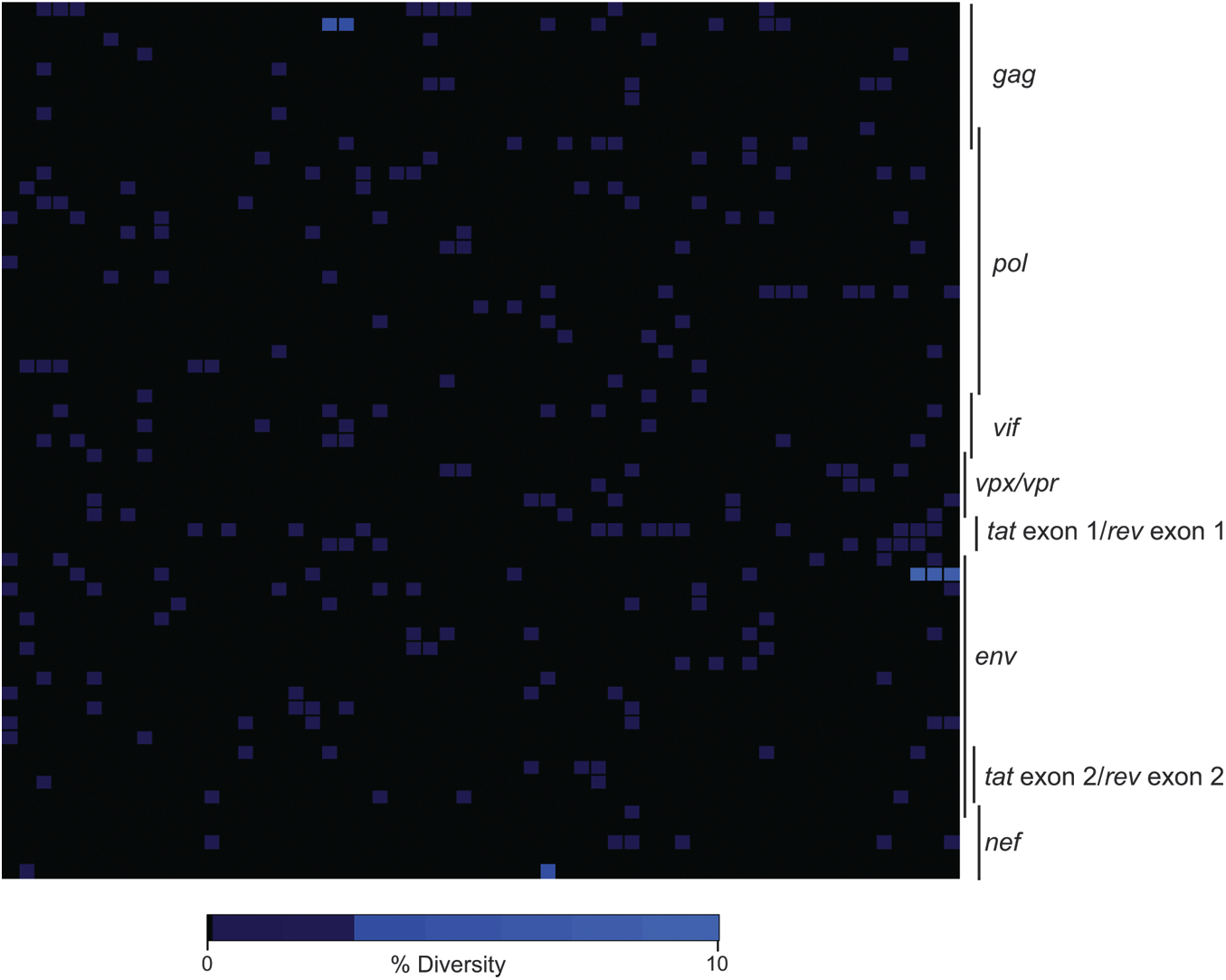
Genome-wide deep sequencing of the SIVmac239 stock used for the IR challenges. Heat map illustration of the level of sequence variation across the SIVmac239 genome detected from 454 deep sequencing. Plotted is the percentage of amino acid diversity at each position across the genome with the first amino acid of Gag located in the top left corner of the grid and the last amino acid of Nef located in the bottom right corner. Completely conserved codons are black, and low-level variant residues (<10%) are colored light blue.
Discussion
In this study, we report the efficacy of a rDNA/rYF17D/rRRV PBB regimen encoding immunodominant SIV epitopes against repeated IR challenges with SIVmac239. Both Mamu-B*08+ Group 1 vaccinees controlled viral replication to levels rarely seen among MHC class I-matched historical control macaques that have been previously infected with SIVmac239. CD8αβ+ T lymphocytes were likely responsible for this impressive outcome as mAb-mediated depletion of these cells resulted in transient increases in viremia. As for the Mamu-A*01+ Group 2 vaccinee r08019, virologic control appeared to involve CD8αα+ lymphocytes and an attenuating deletion in the nef gene, although it is also possible that SIV-specific CD8αβ+ T cells participated in viral containment before the CD8β depletion. Although the mechanism underlying r08019's impressive containment of SIV infection might not be generalizable, the uniqueness of this animal's course of infection illustrates the complexity of lentivirus-host interactions.
The discovery of a nef-deleted SIV variant in r08019 remains a puzzling aspect of this study since this virus was found in all proviral sequences amplified at week 2 PI, but not in the challenge inoculum. Assuming that the low dose SIVmac239 IR challenge regimen used in this study resulted in the transmission of a single virus, we can envision at least three possible explanations for the origin of this nef-deleted mutant. First, despite our inability to detect this deleted form in the SIVmac239 challenge stock, we cannot rigorously rule out the possibility that a minor variant harboring this change is what was transmitted. Indeed, deep sequencing of the inoculum revealed low levels of sequence diversity, probably as a result of propagating the virus on mitogen-activated rhesus macaque PBMC as part of its derivation method. 69 Given the error-prone nature of retroviral reverse transcription, the low frequency SIV variants detected in the stock likely represent byproducts of multiple rounds of viral replication in vitro. Furthermore, our inability to detect the nef-deleted virus in this stock would be in keeping with the technical problems inherent in searching for a rare polymorphism in a highly concentrated sample made almost exclusively of WT genomes.
Second, it is also possible that a reverse transcription error occurred in the first round of viral replication in vivo, resulting in the nef deletion in question. Indeed, short direct repeats are present near the ends of the 18-bp deletion in the SIVmac239 clone (Fig. 6C), similar to what has been observed in other examples of appearance of deletions in nef. 70 These short direct repeats have been associated with the emergence of deletions in retroviral genomes by a mechanism involving template slippage during the reverse transcriptase reaction. 71,72
A third possibility is that the nef deletion was selected for in vivo within the first 2 weeks after r08019 became infected. Although this 2-week time frame might appear too short for the selection of the nef-deleted mutant, there is precedent for immunological pressures resulting in an altered variant virus with similar kinetics. 32,58,73 While the nature of the selection event(s) implicit in this model remains unknown, it probably did not involve T cell responses since r08019 had no detectable Nef-specific T cells in the acute phase (Fig. 7). Alternatively, the nef deletion might have emerged in response to selective pressure imposed by NK cells, potentially by a mechanism involving killer immunoglobulin-like receptor (KIR) recognition of viral epitopes presented by MHC class I molecules. 74 Along these lines, specific aa substitutions in the HIV-1 proteome have been reported to be significantly enriched in infected individuals expressing combinations of KIR and MHC class I alleles, indicating that NK cells can drive HIV-1 escape. 75,76 Given the high epitope density of the core region of Nef, 77 the deleted virus found in r08019 may have been selected for by KIR-expressing NK cells targeting a peptide in that region. Much more extensive analysis will be required to investigate this last possibility.
The use of a new anti-CD8β mAb that selectively eliminates CD8αβ+ T lymphocytes in vivo without the caveat of depleting CD8αα-expressing rhesus macaque NK cells was a key part of this study. A single infusion of this antibody resulted in a surge of SIV replication in the Group 1 macaques but, surprisingly, not in r08019, indicating that this animal did not require an intact CD8αβ+ T cell compartment to maintain control of chronic phase viremia. Importantly, the nef-deleted SIV variant that infected r08019 was clearly replication competent, as evidenced by the sharp rise in VL triggered by the subsequent CD8α depletion. These findings are intriguing and raise the question of what prevented this variant from rebounding in r08019 after the CD8β depletion. Of note, mAb-directed elimination of lymphocytes in tissues can be less efficient than what is observed in blood. Indeed, macaques subjected to the same CD8β255R1 infusion utilized in this study exhibited incomplete depletion of CD8+ T cells in lymph node and duodenal biopsies in the first few weeks after the CD8β255R1 infusion (
Given the increase in CD8αα+ T cell counts after the CD8β depletion, especially in the Group 1 macaque r07032, we also considered the possibility that these cells contributed to control of SIV replication during this period. The physiological role of CD8αα homodimers expressed by T cells is not completely understood. Previous studies in mice have shown that while both CD8αα and CD8αβ isoforms can bind to MHC class I molecules, 82 –84 CD8αβ heterodimers appear to be required for the selection of MHC class I-restricted CD8+ T lymphocytes in the thymus. 50 Furthermore, the CD8αα molecule has been described as a marker for T cell activation and is primarily expressed by mucosal T cells. 51,85,86 Interestingly, human CD8αα+ T cells residing in genital skin have been recently implicated in the containment of herpes simplex virus-2 reactivation, 87 indicating that these cells participate in defense against persistent viral infections. Although CD8αα+ T cells might be part of a unique, regionally specialized, lineage of T lymphocytes, it is not clear if the peripheral CD8αα+ T cells identified in this study are examples of such cells. Alternatively, they could have comprised preexisting CD8αα+CD8αβ+ T cells in PBMC that internalized their surface CD8αβ heterodimers upon ligation of the anti-CD8β mAb. A detailed analysis of the T cell receptor clonotypes, Ag specificities, and tissue distribution of CD8αα+ and CD8αβ+ T cells before and after depletion of CD8αβ+ lymphocytes would have been needed to address this possibility. Nevertheless, in light of the recent identification of tissue-resident human CD8αα+ T cells endowed with potent antiviral activity, 87 characterizing the role of these unconventional T lymphocytes during lentivirus infections may lead to new strategies for eliciting cellular immunity against HIV.
In conclusion, in this study we show that SIV-specific CD8+ T cells elicited by a rDNA/rYF17D/rRRV PBB regimen afforded significant control of SIVmac239 replication in 3/4 vaccinees. Since vaccine-induced CD8+ T cells in these animals were focused on a single immunodominant SIV epitope, we are currently exploring if expanding the CD8+ T cell response against other viral Ags can improve vaccine efficacy. We also explored the basis for the unusual control of SIVmac239 replication manifested by one vaccinated macaque and showed that, surprisingly, this animal harbored a nef-deleted mutant as early as 2 weeks PI. Subsequent CD8 depletions suggested that replication of this SIV variant may have been contained by CD8αα-expressing lymphocytes, warranting further investigation of the antiviral capacity of NK cells in vivo. While we could not pinpoint the origin of the nef-deleted virus, three possible explanations involving both stochastic and selective events were presented based on the available data. Collectively, these data provide additional insights into the full spectrum of virologic control of lentivirus infection.
Footnotes
Acknowledgments
The authors are grateful to Brandon Keele for helpful comments on this article and to Dr. Elizabeth Connick for quantifying SIV-producing cells in lymph node biopsies by in situ hybridization. The authors also thank Rebecca Shoemaker, Nicholas Pomplun, Jessica Furlott, Randy Fast, Kelli Oswald, Marlon Veloso de Santana, and William Lauer for technical support. The authors also wish to acknowledge Eric Peterson, Kristin Crosno, and Kevin Brunner for providing excellent care of the rhesus macaques used in this experiment; Teresa Maidana Giret for confirming the MHC class I genotype of the monkeys in this study; Leydi Guzman for administrative assistance; and Christopher Parks for facilitating the electroporated rDNA vaccinations used in this study. This work was funded by Public Health Service grant R37AI052056 to D.I.W. and was supported, in part, by federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E and from the National Institute of Allergy and Infectious Diseases through P01 AI074415 (T.M.A.). The funders had no role in study design, data collection, and interpretation or the decision to submit the work for publication. The mAbs used in the in vivo lymphocyte depletions were produced by the NIH Nonhuman Primate Reagent Resource (OD010976 and HHSN2722001300031C, K.A.R.).
Disclaimer
TA's spouse was an employee of Bristol-Myers Squibb, which has a focus in Virology, specifically treatments for hepatitis B and C and HIV/AIDS. TA's spouse no longer works for BMS and only retained a small stock interest in the public company. TA's interests were reviewed and managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Sequence Data
All reads have been deposited to the NCBI Sequence Read Archive under accession number SRP016012.
Author Disclosure Statement
No competing financial interests exist.