
Abstract
CRF07_BC was originally formed in Yunnan province of China in 1980s and spread quickly in injecting drug users (IDUs). In recent years, it has been introduced into men who have sex with men (MSM) and become the most dominant strain in China. In this study, we performed a comprehensively phylodynamic analysis of CRF07_BC sequences from China. All CRF07_BC sequences identified in China were retrieved from database. More sequences obtained in our laboratory were added to make the dataset more representative. A maximum-likelihood (ML) tree was constructed with PhyML3.0. Maximum clade credibility (MCC) tree and effective population size were predicted by using Markov Chains Monte Carlo sampling method with Beast software. A total of 610 CRF07_BC sequences coving 1,473 bp of the gag gene (from 817 to 2,289 according to HXB2 calculator) were included into the dataset. Three epidemic clusters were identified; two clusters comprised sequences from IDUs, while one cluster mainly contained sequences from MSMs. The time of the most recent common ancestor of clusters that composed of sequences from MSMs was estimated to be in 2000. Two rapid spreading waves of effective population size of CRF07_BC infections were identified in the skyline plot. The second wave coincided with the expanding of MSM cluster. The results indicated that the control of CRF07_BC infections in MSMs would help to decrease its epidemic in China.
H
In China, the proportion of HIV infections caused by CRFs is even higher. According to a comprehensive study on distribution of HIV subtypes in China, more than 80% HIV new infections were due to CRFs. 8 CRF07_BC was the most dominant subtype, which accounted for 35.5% of all new HIV infections. 8 CRF07_BC is believed to be formed in the Yunnan province of China, and then spread to Sichuan, Gansu, and finally to Xinjiang province, along the drug trade route. 9 –11 After that, CRF07_BC spread quickly in injecting drug users (IDUs) and became dominant in many provinces. In some areas, for example in Sichuan province, CRF07_BC even accounted for more than 90% of HIV infections. 12
In the last decade, the CRF07_BC strain was frequently found in men who have sex with men (MSM). 13,14 MSMs are the population most at risk of HIV infection in virtually all regions other than sub-Saharan Africa, especially in industrialized countries. In China, the proportion of HIV infections due to homosexual contact has increased dramatically in recent years. Before 2005, MSMs only accounted for 0.3% of new HIV infections in the whole country. In 2011, the proportion increased to 13.7%. Therefore, we hypothesized that the introduction of CRF07_BC into MSMs might cause its rapid spreading. The purpose of this study was to investigate the effect of CRF07_BC epidemic in MSMs on total CRF07_BC prevalence in China.
All HIV-1 CRF07_BC sequences isolated in China in Los Alamos HIV sequence database (
Finally, a total of 605 HIV-1 CRF07_BC sequences spanning about 1,473 bp of the gag gene (from 817 to 2,289 according to HXB2 calculator) were included into the dataset for further analysis. The isolating time of strains spanned from 1997 to 2013. Among them, 180 sequences were retrieved from the Los Alamos HIV sequence database and the other 430 sequences were from our own cohort. The areas of those strains cover 12 of 31 provinces in China (Fig. 1). 78.65% sequences were from IDUs, while 5.09% sequences were from MSMs.
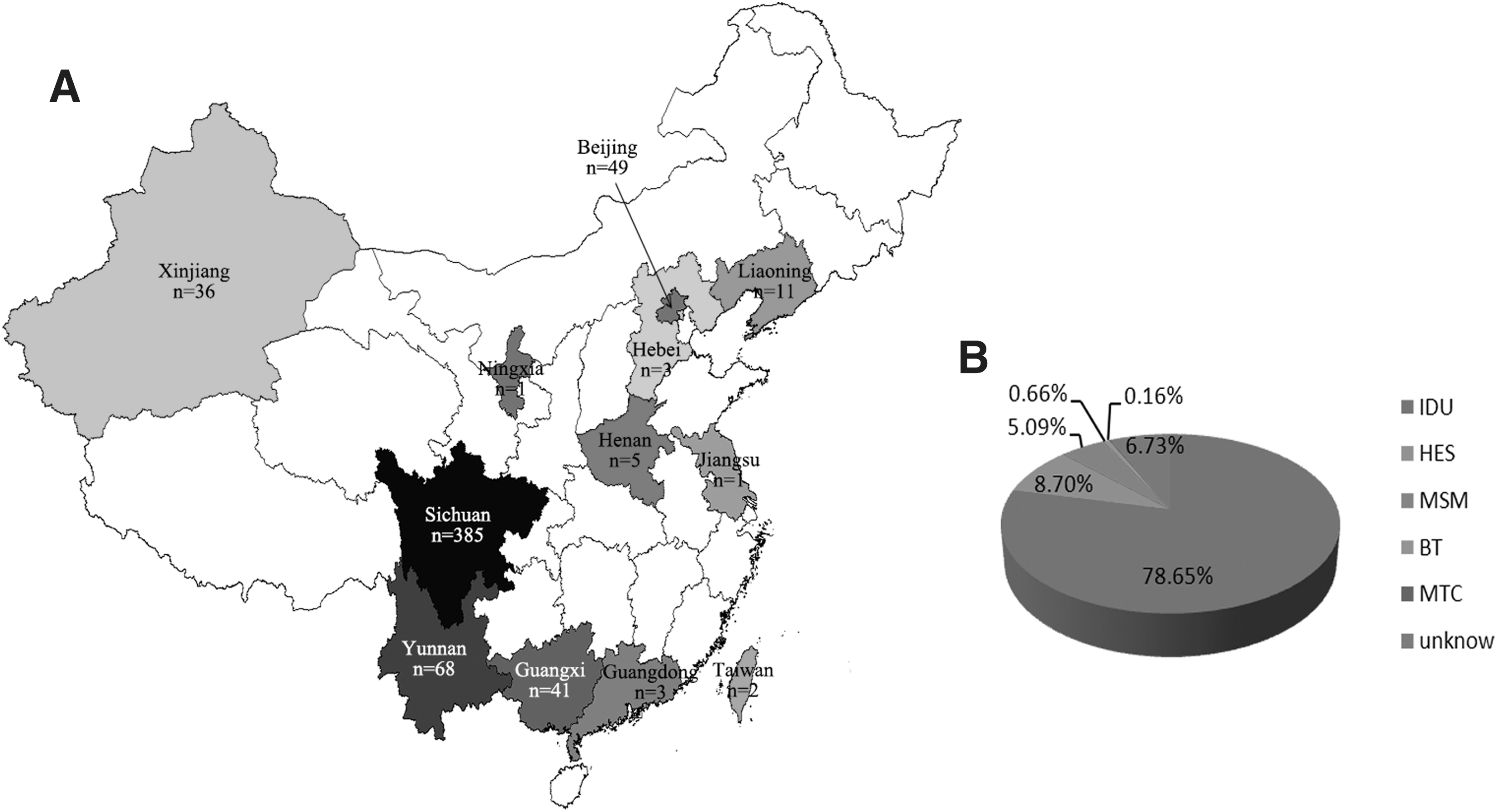
Distributions of CRF07_BC strains used in this study based on geographical locations or risk factors.
All sequences were aligned using Muscle software, manually edited when necessary. Four subtype C reference sequences were added into the dataset as out groups. A maximum-likelihood tree was constructed by using GTR+I+G (General Time Reversible+Invariant+Gamma) nucleotide substitution model, which was selected by jModelTest. Subtree pruning and regrafting (SPR) was used for tree searching. The confidence values of tree branches were tested by using approximate likelihood-ratio test, which was accurate and fast enough for a large dataset. The final tree was edited and displayed using FigTree v1.4.2. Epidemic clusters were defined as clades containing more than 10 sequences with a branch support value higher than 90%.
In the maximum-likelihood tree containing 609 sequences (Fig. 2A), three epidemic clusters were identified with confidence value higher than 90% in the maximum-likelihood (ML) tree. Clusters I and III were mainly composed of sequences from the IDU population (more than 95% sequences), and Cluster II was predominantly composed of sequences from the MSM population. For area distribution, cluster I mainly contained sequences from Sichuan province, cluster III mainly contained sequences from Guangxi provinces, while cluster II was composed of sequences from comprehensive areas. Since Sichuan and Guangxi provinces were both along the drug traffic road, it is reasonable to find cluster I and III separately, considering that CRF07_BC is mainly circulating in IDUs. 12 A few sequences in cluster I and III were isolated from patients infected through sexual transmission, supposing that CRF07_BC strains might have gone out of IDUs into general populations. Different to cluster I and III, cluster II contained mainly sequences from MSMs. Patients with viruses in cluster II were from a wide geographic region, suggesting that MSMs often travel large distances in comparison to IDUs.

Phylogenetic tree analysis and Bayesian skyline analysis.
The rapid accumulation of HIV genetic variation due to short generation times and high mutation rates makes it possible to investigate its epidemic spread basing on viral phylodynamics analysis. Bayesian Markov Chain Monte Carlo evolutionary analysis was further fulfilled step by step as described previously to explore the expansion of CRF07_BC strains in China.
15
First, the dataset was balanced to made it representative and maximize the “clock-likeness.” Second, RDP
16
software was used to test and remove all of intra-subtype recombinant sequences. Third, Path-O-Gen (
The general time reversible (GTR) model was selected as the most appropriate nucleotide substitution model for this dataset by using jModelTest. 17 Nonparametric model (Bayesian Skyline Plot with strict clock model) was initially selected to infer the demographic information within the database using BEAST v1.7.5. 18 Then, several parametric demographic models, including constant population size, exponential growth, logistic growth, expansion growth and Bayesian skyline were compared by the Bayesian factor, and Bayesian skyline was used as the final model. The model was run to 1 × 109 generations, sampling every 1000th generation. The first 10% of the output was used as a burn-in. Convergence of the estimates was evaluated with generation versus log probability plots in Tracer v.1.519 using an Effective Sample Size >200. A maximum clade credibility tree was generated using TreeAnnotator in BEAST, which was further edited with Figtree v1.3.1. 18 To infer the demographic history of CRF07_BC in China, the nonparametric reconstruction of the epidemic history with appropriate confidence limits was fulfilled, showing change in the effective number of infections.
The Bayesian analysis shows that the time the most recent common ancestor (tMRCA) of CRF07_BC was predicted was in 1989, which is similar as deciphered by Tee et al. in 2008. 11 The two clusters come from the IDU population identified in the ML tree, also presented in the Bayesian tree with high possible posterior value. Also, the tMRCA of cluster I and cluster III was estimated to be in 1994 [95% highest posterior density (HPD) 1991–1997] and 2001 (95% HPD 1998–2003). The tMRCA of cluster II was estimated to be in 2000 (95% HPD 1997–2003), which heralded the epidemic of CRF07_BC infections through ongoing intra-MSM transmissions.
Two growth waves were observed in the effective population size of CRF07_BC infection in skyline plot, suggesting that there are two exponential growth at 1991 and 2003, respectively (Fig. 2A). When sequences in cluster II were deleted from the dataset, the second wave of effective population size in skyline plot diminished accordingly, suggesting that the second wave of CRF07_BC spreading was caused by its prevalence in MSMs.
HIV-1 CRF07_BC is the main subtype in China. In the study, a total of 605 HIV-1 CRF07_BC sequences between 1997 and 2013 were collected to construct a maximum-likelihood tree. Three clusters with more than 10 sequences and confidence value higher than 90% in the ML tree were identified, which could also be identified in the maximum clade credibility (MCC) tree with high possible posterior value. Two rapid growth period of effective population size were observed in the skyline plot, suggesting two rapid spread of the CRF07_BC.
CRF07_BC is the most dominant strain prevalent in China; understanding its epidemic history will be helpful for the control of HIV infections. In this study, we identified two rapid spreading waves in the history of CRF07_BC epidemic. The first rapid spread of CRF07_BC was observed before 1997 in the skyline plot. It should be caused by its prevalence in IDUs after its initial infection. After 1997, the growth rate of CRF07_BC in China slowed down until 2003, suggesting that the infection of CRF07_BC in IDU population got closer to saturation. The second sharp increase of EPS in the skyline plot was observed in 2003, which is similar to the time when CRF07_BC was introduced into MSMs. Deletion of sequences from MSMs disappeared the growth curve, which proved that CRF07_BC epidemic in MSMs caused its second spreading wave in China. The results strongly supposed that MSMs should be the emphasis of interventions for HIV control.
Sequence Data
The gene sequences were deposited in the GenBank with the following accession numbers: KP234525-KP234916 and KY847803-KY847856.
Footnotes
Acknowledgments
This work was supported by the National Key S&T Special Projects on Major Infectious Diseases (grant no. 2012ZX10001-002) and the National Natural Science Foundation of China (no. 81273137).
Author Disclosure Statement
No competing financial interests exist.