
Abstract
HIV-1 viremia has been shown to induce several phenotypic and functional abnormalities in natural killer (NK) cells. To assess immune defects associated with HIV viremia, we examined NK cell function, differentiation status, and phenotypic alterations based on expression of inhibitory and activating receptors on NK cells in HIV-1 subtype C chronically infected participants from Durban, South Africa. NK cell phenotypic profiles were characterized by assessing sialic acid-binding immunoglobulin-like lectin-7 (Siglec-7), NKG2A, and NKG2C markers on frozen peripheral blood mononuclear cells from viremic, antiretroviral therapy (ART)-naive HIV-1 chronically infected participants (n = 23), HIV-1 chronically infected participants who had been on combination antiretroviral therapy (cART) for at least 12 months (n = 23) compared with healthy donors (n = 23). NK cell differentiation was assessed by measurement of killer immunoglobulin receptor (KIR) and NKG2A expression; CD57 and CD107a measurements were carried out in HIV viremic and healthy donors. All phenotypic and functional assessments were analyzed by using multicolor flow cytometry. HIV-1-infected participants displayed greater frequencies of the CD56−CD16+ (CD56negative) NK cell subset compared with healthy donors (p < .0001). Downregulation of Siglec-7 and NKG2A and upregulation of NKG2C were more pronounced in the CD56negative NK cell subset of viremic participants. The CD56negative subset demonstrated a differentiated (KIR+NKG2A−) phenotype with reduced CD57 expression and lower degranulation capacity in HIV-1-infected participants compared with healthy donors. HIV-1 infection induces the expansion of the CD56negative NK cell subset marked by altered receptor expression profiles that are indicative of impaired function and may explain the overall NK cell dysfunction observed in chronic HIV-1 infection.
Introduction
N
NK cells express an array of activating and inhibitory receptors, and it is the balance between these receptors that mediates the functional activity of these cells. 15,16 One of the proposed mechanisms used by HIV-1 to efficiently evade NK cell immune responses is through downregulation of activating NK cell receptors, resulting in impairment of NK cell activities. 11,17 Among the frequently studied receptors are the killer immunoglobulin receptors (KIRs) and NKG2, C-type-lectin family, 18 well established to play an essential role in NK cell education that renders the cells competent or hypo-responsive. 9,19 –22 A report by Beziat et al. suggested a reclassification of NK cell differentiation based on a stepwise decrease of NKG2A and acquisition of KIRs and demonstrated that the CD56dim NKG2A−KIR+ subset is the most differentiated, is capable of lysing target cells, and also has a progressive increase in the expression of CD57. 23 Similarly, CD57 has been shown to be highly expressed on NK cell subsets with a mature phenotype, marking NK cells that have been clonally expanded by infections. 24 HIV-1 infection has also been shown to mediate a switch from the inhibitory receptor NKG2A to an activating receptor NKG2C on NK cells, resulting in an imbalance between NKG2A- and NKG2C-expressing cells. 25 –27 On the other hand, sialic acid-binding immunoglobulin-like lectin-7 (Siglec-7) has been reported as a sensitive marker for identifying and tracking NK cell phenotypic changes and impaired function during different stages of HIV-1 infection 28 ; however, establishment of its utility as a marker of NK cell dysfunction requires investigation in other cohorts.
Overall, there is paucity of data and, thus, a need to identify factors that may lead to NK cell dysfunction in chronic HIV-1 infection. In particular, identification and tracking of phenotypic alterations in various NK cell subsets and their impact on overall NK cell function during chronic infection in African cohorts are lacking. We used multi-parameter flow cytometry to examine the phenotypic differences on NK cells based on surface expression of Siglec-7, NKG2A, and NKG2C among black Africans with either progressive or treated HIV infection compared with HIV-uninfected donors. We also assessed the differentiation status (based on KIR and NKG2A expression) and the functional significance of these alterations mediated by HIV infection on the CD56−CD16+ (CD56negative) NK cell subset in progressive clade C HIV-1-infected individuals.
Materials and Methods
Study participants
HIV-1 subtype C chronically infected participants were recruited from the Sinikithemba cohort in Durban, KwaZulu-Natal in South Africa; the characteristics of this cohort have been previously described by Wright et al. 29 Healthy donors were recruited from the Masibambisane cohort of pregnant mothers aged 16–22 years who frequently visited the maternity clinic at the Prince Mshiyeni Hospital, Durban. All volunteers gave informed consent to participate, and the studies received ethical clearance from the University of KwaZulu-Natal Biomedical Research Ethics Committee, reference numbers (E028/99 and BF168/09). A total of 69 participants were divided into 3 study groups as follows: antiretroviral therapy (ART)-naive individuals with progressive HIV-1 infection (n = 23), HIV-1-infected individuals who had been on combination (cART) for at least 12 months (n = 23), and lastly, healthy HIV-negative individuals (n = 23). Exclusion criteria included co-infection with tuberculosis. Clinical characteristics of the study participants are shown in Table 1.
cART, combination antiretroviral therapy; IQR, interquartile range.
Flow cytometry
Cryopreserved PBMCs were thawed and washed with R10 (RPMI medium containing 10% heat-inactivated fetal calf serum, 1%
Viral load and CD4 count measurements
Plasma viral loads were measured by using either the Roche Amplicor Monitor assay with a detection limit of 400 HIV-1 RNA copies/ml plasma or the Roche Ultrasensitive assay with a detection limit of 50 RNA copies/ml plasma, according to the manufacturer's instructions. CD4 counts were determined from fresh whole blood by using Tru-Count technology and analyzed on a four-color flow cytometer (Becton Dickinson) according to the manufacturer's instructions.
Data analyses and statistics
Data of scatterplots from the flow cytometer (FCS files) were analyzed by using FlowJo software version 9.5.5 (Tree Star, Ashland, OR). Differences between the three studied groups were examined by using Kruskal–Wallis test with Dunn's post-test followed by Mann–Whitney U test. Frequencies of cells expressing measured receptors/markers and cytokines between specified NK cell subsets in healthy donors and HIV-infected (either viremic or treated) groups were compared by the Mann–Whitney U test by using GraphPad Prism version 5.0 software (GraphPad Software, Inc., La Jolla, CA). Differences between groups were considered statistically significant at p < .05 level.
Results
HIV-1 infection is associated with expansion of the CD56−CD16+ NK cell subset
We examined phenotypic differences in NK cells based on their surface expression of Siglec-7, NKG2A, and NKG2C as well as assessed differentiation status by KIR and NKG2A expression by using a model described by Beziatet al. 23 NK cells were identified as viable CD3−CD14−CD19− lymphocytes, and total NK cells were defined by CD56 and CD16 surface marker expression (Fig. 1A). NK cells were further classified, based on relative expression of CD56 and CD16 markers, into three major subsets: the CD56+CD16−(bright) subset, the cytotoxic CD56+CD16+(dim) subset, and the “pathologic” CD56−CD16+(negative) subset, 8 (Fig. 1B). The differences in the expression of the phenotypic markers listed earlier as well as differentiation status were assessed in the total NK cell population (defined here as combined CD56+CD16−, CD56+CD16+, and CD56−CD16+ subsets) and in the different subsets specified by KIR and NKG2A (Fig. 1C).
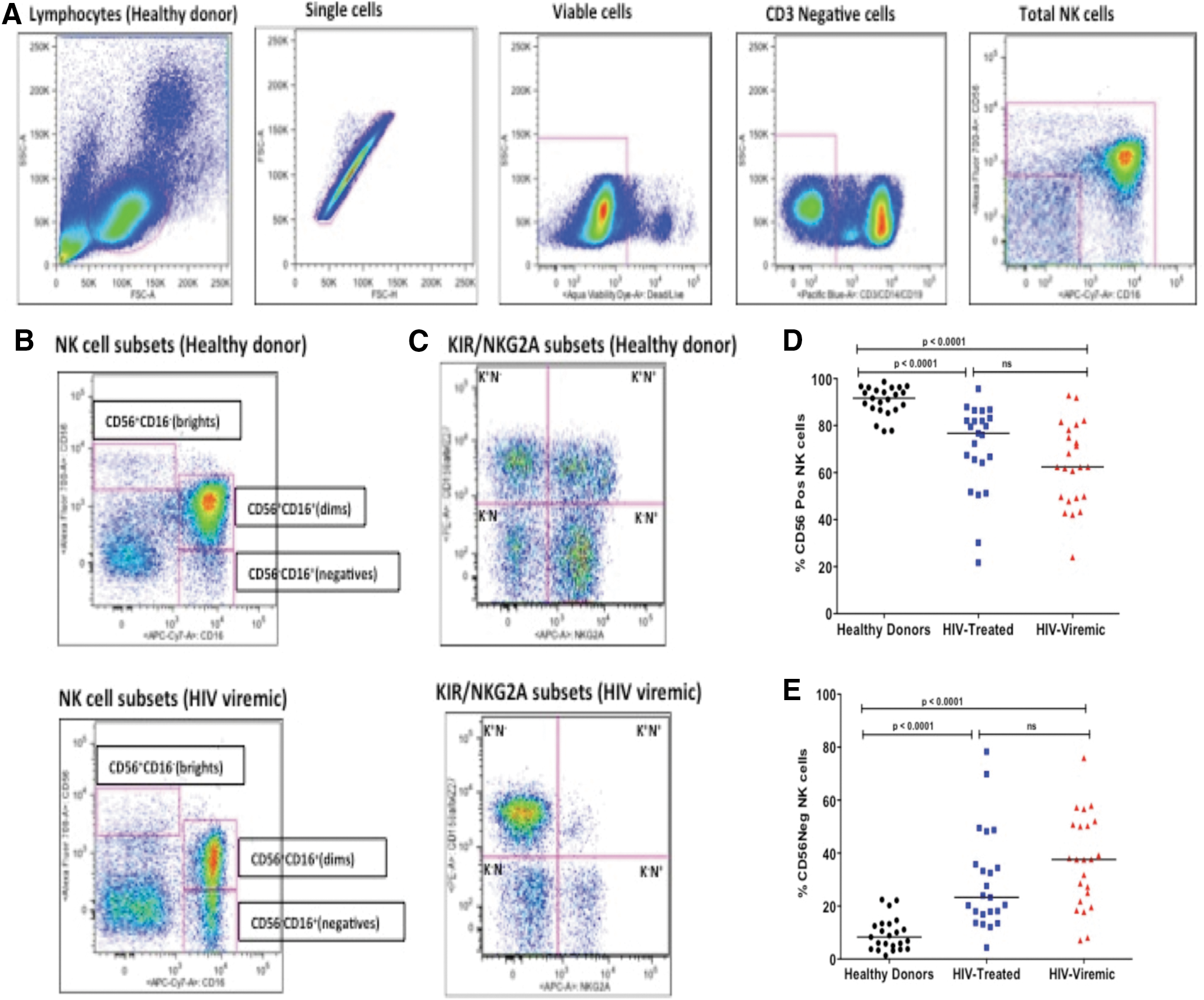
Representative NK cell gating strategy used to identify total NK cell (CD56brights, CD56dims and CD56negatives combined) population
Our data indicate that the total frequency of CD56+ NK cells (inclusive of both CD56bright and CD56dim NK cell populations) was higher in healthy donors compared with either treated HIV-1-infected subjects or those with progressive infection (Fig. 1D). However, no significant differences were noted in the CD56bright population between healthy donors, therapy-naive, and treated HIV-infected individuals (data not shown). In contrast, the frequency of CD56negative NK cells was greater in therapy-naive HIV-1 chronically infected individuals and individuals on cART when compared with healthy donors (p < .0001 for both comparisons, Mann–Whitney; Fig. 1E). The median frequency of the CD56negative NK cells in healthy participants was 8.3, interquartile range (IQR: 3.8–13.3) whereas the median of treated HIV-infected participants was 23.3, IQR (16.9–35.8) and a median of 37.6, IQR (21.8–50.8) was observed for the viremic HIV-infected participants (depicted in Fig. 1E).
Reduced Siglec-7 and NKG2A and expansion of NKG2C expression in HIV-1-infected participants
To further characterize the impact of HIV infection on NK cells in this cohort, we assessed Siglec-7, NKG2A, and NKG2C expression profiles on different NK cell subsets in either treated or progressive chronic HIV-1 infection compared with healthy donors. The frequency of Siglec-7 expression was significantly lower in total NK cells (all NK cell subsets, including CD56negative cells) in therapy-naive and treated HIV-1-infected groups when compared with healthy donors (p < .0001 and p = .004 respectively, Mann–Whitney, Fig. 2A). Further analysis based on NK cell subsets demonstrated that this lower frequency of Siglec-7+ NK cells was also observed in CD56negative NK cell subsets of individuals with untreated HIV infection when compared with healthy donors (Fig. 2B). A similar pattern was observed in CD56+ (consisting of CD56bright and dim populations) NK cells, where significantly lower frequencies of Siglec-7+ NK cells were noted in individuals with progressive infection compared with healthy donors (data not shown).
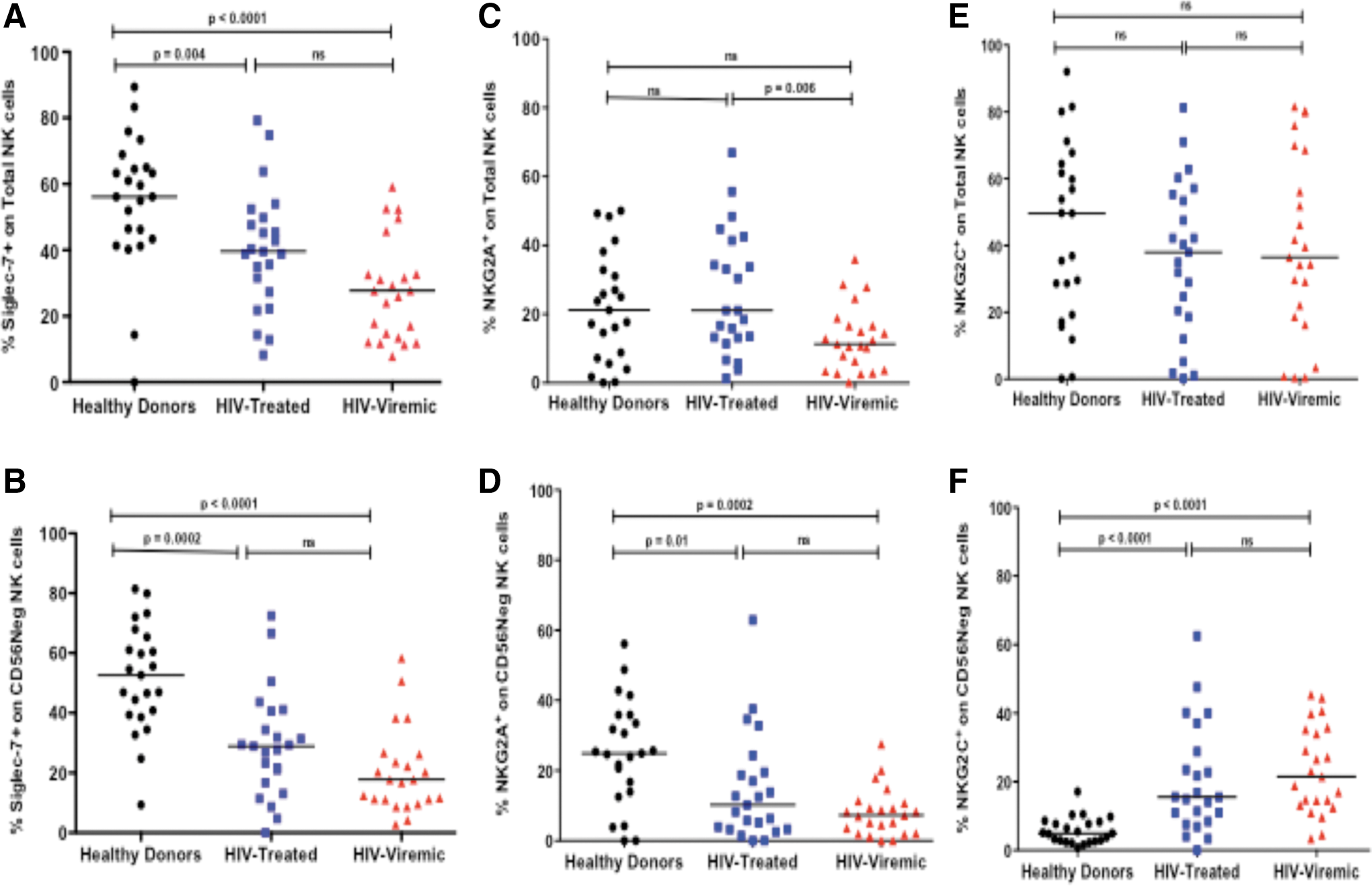
Frequency of Siglec-7
In total NK cells, the frequency of NKG2A+ NK cells was lower in HIV viremic subjects compared with treated subjects. However, no difference in NKG2A frequency was distinguished in total NK cells when comparing healthy donors with either of the HIV-1-infected groups. We noted a similar trend to that of Siglec-7 in the CD56negative subset where lower frequencies of NKG2A+CD56negative NK cells in HIV-1-infected individuals compared with healthy donors were observed (Fig. 2C, D). In contrast, the frequency of NKG2C+ NK cells was higher in HIV-infected groups compared with healthy donors in the CD56negative subset, but not in total NK cells (Fig. 2E, F). Taken together, these findings suggest that expansion of the CD56negative NK cell subset during chronic HIV infection may lead to phenotypic variations that are not always detected when examining the total NK cell population.
CD56negative NK cells from chronically infected individuals are terminally differentiated (KIR+NKG2A−) with reduced CD57 expression
Given the earlier findings on the expansion of the CD56negative subset and phenotypic alterations observed in this subset, we next assessed the differentiation status of the CD56negative NK cell subset in healthy donors and chronically HIV infected subjects based on KIR and NKG2A as proposed by Beziat et al. 23 The authors previously defined a new model of terminal differentiation in NK cells based on a stepwise decrease of NKG2A and acquisition of KIRs as a determinant of the functional fate of NK cells. Our results confirm significantly lower frequencies of NKG2A-expressing NK cells (KIR−NKG2A+ and KIR+NKG2A+; p = .0005, p = .004; Mann–Whitney) in chronically infected individuals compared with healthy donors and expansion of the terminally differentiated KIR-expressing NK cells lacking NKG2A expression (KIR+NKG2A−, p = .001, Mann–Whitney Fig. 3A). Interestingly, the terminally differentiated (KIR+NKG2A−) CD56negative NK cell subset was the only subset found to have a reduced expression of CD57 in chronically infected subjects when compared with healthy donors (p = .003, Mann–Whitney; Fig. 3B). No differences were observed in CD57 expression in other KIR/NKG2A CD56negative NK cell subsets.
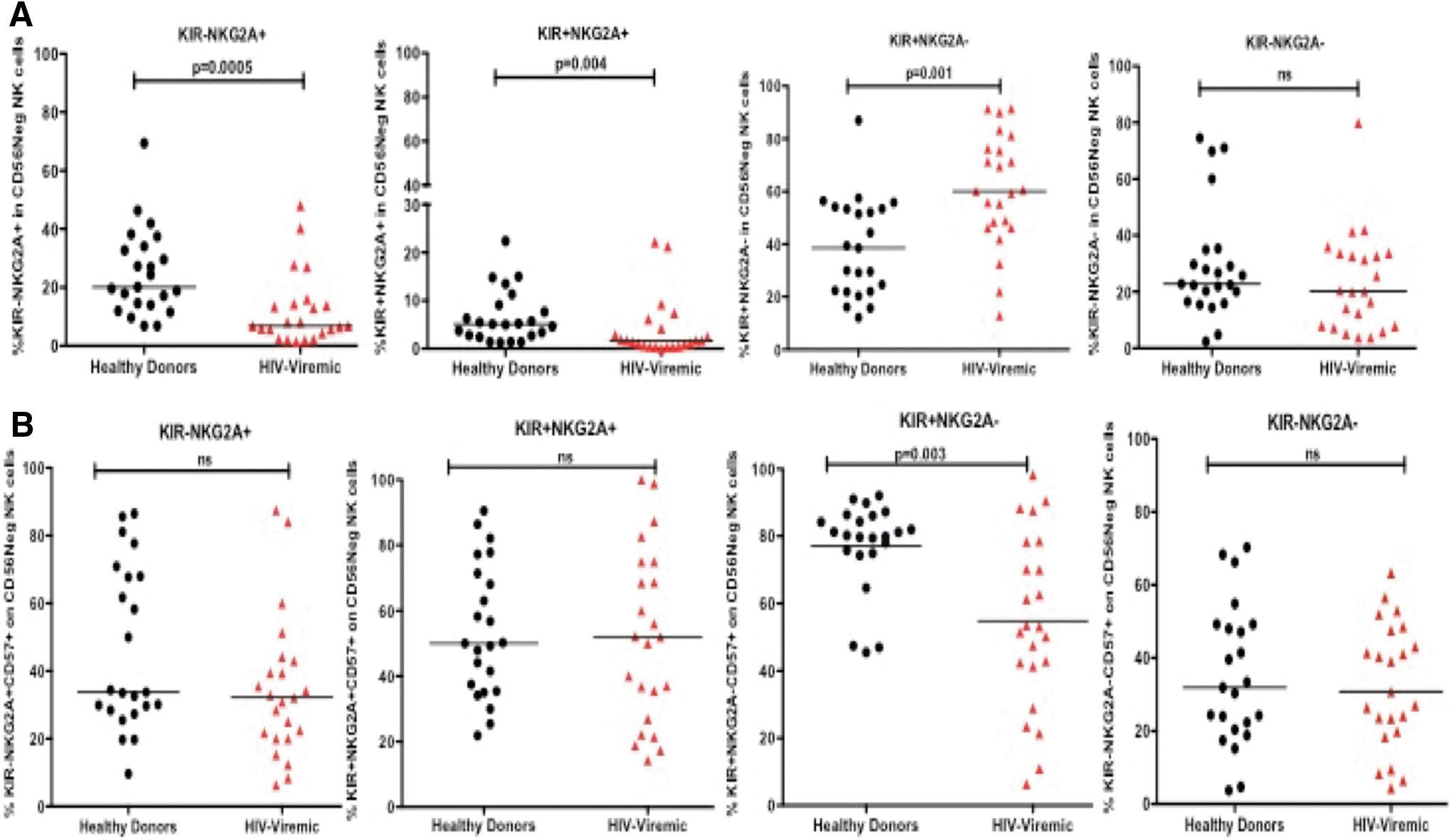
Frequencies of KIR/NKG2A combination profiles in the CD56negative NK cell subset
CD56negative NK cell subset of HIV-1 infected participants has impaired NK cell degranulation capacity
We next assessed the impact of the altered phenotype noted on the CD56negative NK cell subset on NK cell function by investigating the ability of NK cells to degranulate (CD107a expression) after overnight stimulation with K562 target cells. We first evaluated the overall ability of KIR/NKG2A CD56negative NK cell subsets to degranulate and found that the dominant subset during HIV-1 infection, that is, the terminally differentiated (KIR+NKG2A−) NK cells had reduced degranulation capacity (p = .002; Mann–Whitney; Fig. 4A). Similarly, the other NKG2A lacking subset (KIR−NKG2A−) also showed a trend of a lower degranulation capacity (p = .06; Mann–Whitney), whereas the NKG2A-expressing subset lacking KIR expression (KIR+NKG2A−) showed no significant differences in CD107a expression in chronically HIV-1-infected subjects with progressive infection compared with healthy donors (Fig. 4A). We further assessed K562-induced degranulation in the abnormal CD56negative NK cell subset and noted a reduction in the frequency of CD107a expression in HIV-infected participants compared with healthy donors (p = .03; Mann–Whitney, Fig. 4B). Taken together, our data demonstrate that the CD56negative population that expands during chronic infection may represent an impaired NK cell population during chronic HIV-1 infection.
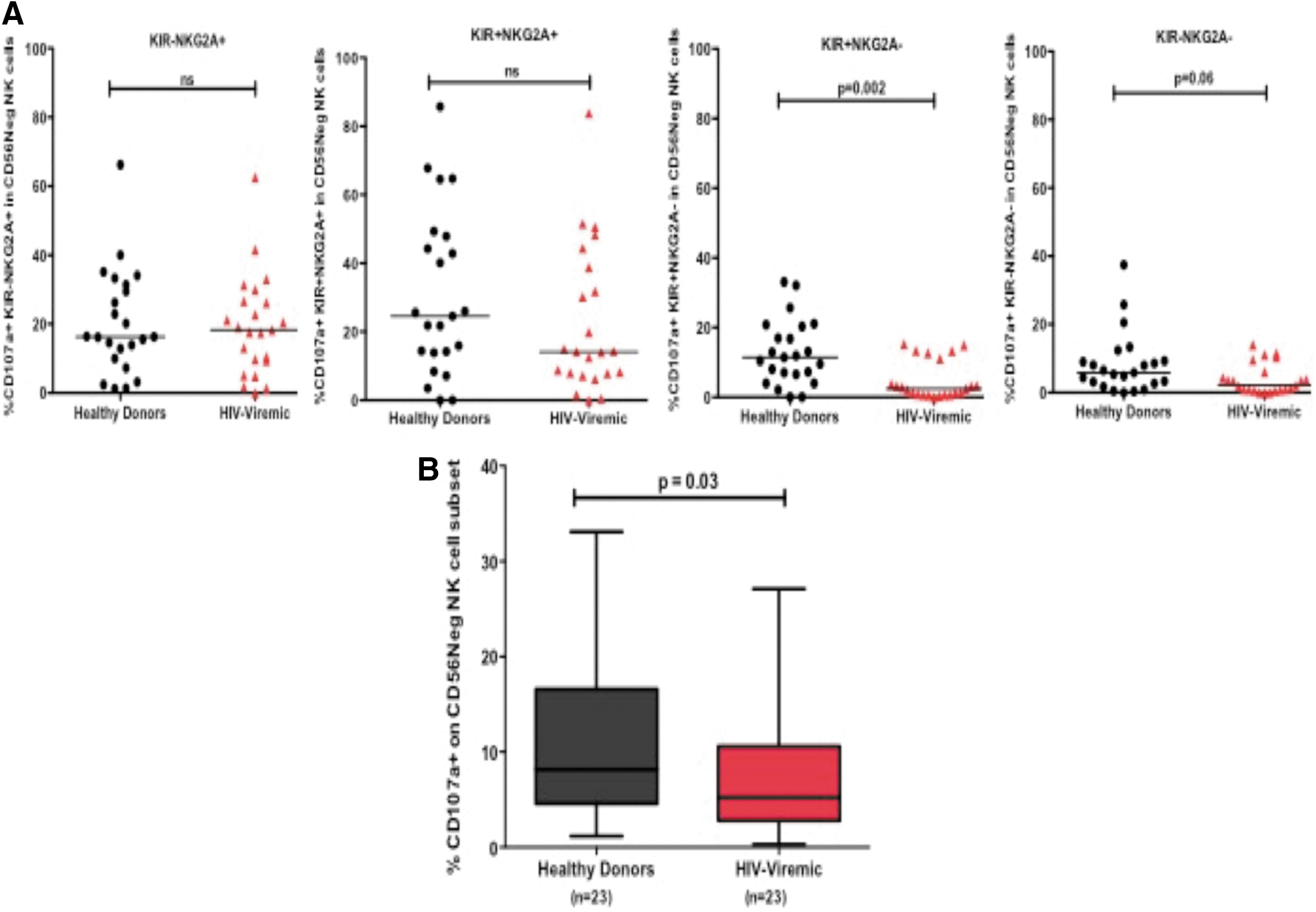
Assessment of degranulation (CD107a expression) by KIR/NKG2A subsets on CD56negative NK cells
Discussion
Several studies have implicated HIV-1 viremia as the main factor behind differences in NK cell subsets and NK cell dysregulation during HIV infection. However, very few of these studies have reported the factors behind this dysfunction and impact on disease in HIV-1 clade C infection, the subtype that is responsible for most deaths in southern African countries. Examination of these dynamics in clade C infected Zulu/Xhosa individuals from Durban, South Africa, demonstrates an expansion of the previously described “pathologic” CD56negative NK cell subset in HIV-1 chronically infected participants with progressive infection compared with healthy donors or individuals undergoing ART and further describes markers expressed by the CD56negative NK cell subset. Second, we show that lower frequencies of Siglec-7+ and NKG2A+ inhibitory NK cell receptors were accompanied by elevated expression of the activating receptor NKG2C+ NK cells in the CD56negative NK cell subset of HIV-1-infected participants with progressive infection. We also demonstrate that the CD56negative NK cells had a terminally differentiated phenotype (defined by being KIR+NKG2A−) with reduced CD57 expression. Lastly, this abnormal and differentiated phenotype was associated with a lower degranulation capacity of the CD56negative NK cells, in HIV-infected individuals compared with healthy donors.
Since first reported, 31 the appearance of the CD56negative NK cell subset has been confirmed in chronic HIV-1 infection by several other studies and has been found to be associated with decreased degranulation and cytokine secretion and selective depletion of the CD56dim NK cell subset. 32 –34 Our data concur with these studies in demonstrating that expansion of the CD56negative NK cell subset coincided with a substantially lower CD56dim (CD56+CD16+) NK cell subset and that 12 months of antiretroviral treatment was not sufficient to restore CD56 expression and function in NK cells to levels observed in healthy donors.
Although a number of studies have assessed Siglec-7, NKG2A, and NKG2C receptor heterogeneity in total NK cell populations, we were particularly interested in differences within individual NK cell subsets, particularly the CD56negative population that was common in HIV-viremic individuals. Our findings display similar observations as seen in total NK cells in previous studies and reveal pronounced differences in the CD56negative NK cell populations. In contrast, higher NKG2C expression was only observed in the CD56negative population and not in total NK cells, demonstrating the importance of assessing receptor expression by subsets. Siglec-7 has been recently reported as a sensitive marker for identifying and tracking NK cell phenotypic changes associated with impaired NK cell function during different stages of HIV-1 infection. 28 Our findings are in line with those described by Brunetta et al. in European cohorts 28 ; here, we also established that African individuals with high HIV-1 levels had lower frequencies of Siglec-7+ NK cells, but expression differences were less pronounced during controlled viremia in antiretroviral-treated individuals. Similarly, as shown in our study, a decrease in NKG2A, a key inhibitory NK receptor, has been described by others. 11 Elevated NKG2C expression, an activating NK cell receptor specific to HLA-E, has been linked to co-infection with human cytomegalovirus (HCMV) 35,36 ; lack of NKG2C+ NK cells has been reported in HCMV seronegative people or present at low levels in individuals with low HCMV IgG titers. 25,37 All participants in our study groups were positive for HCMV-specific IgG (data not shown), and, as such, suggest that HCMV is not the primary factor responsible for high NKG2C expression in the HIV-infected group. Rather factors independent of HCMV co-infection may account for these NKG2C differences in NK cells and warrant further investigation. Nonetheless, our data support previous studies in demonstrating altered NK cell subsets in chronic HIV-1 infection, that is, lower expression of inhibitory receptors and higher expression of activating receptors in comparison to healthy donors. 11,17,38 We also characterized these cells by differentiation status, the results of which indicate that phenotypic alterations have an impact on the overall function of the CD56negative subset in the African population with clade C HIV-1 infection.
The CD56negative characteristics reported in our study are not unique to our cohorts; reduced NKG2A, CD57, Siglec-7, and high KIR expression has been previously reported in non-African cohort settings. 39 Interestingly, many of these variations are also characteristic of CD56dim NK cells, raising questions of CD56negative NK cell origin, which require future clarity. It remains to be determined whether the CD56negative NK cells represent the terminally differentiated NK cells subset or arise from a mixed population of mature CD56dim NK cells with altered characteristics. 39 We speculate that the latter is likely possible as suggested by the KIR+NKG2A− and low CD57 expression phenotype also noted on the CD56dim subset (data not shown). CD57 on NK cells represent a state of NK cell maturation and higher cytotoxic potential. 13,40 It is, thus, possible that CD57 down-modulation on the terminally differentiated KIR+NKG2A− CD56negative NK cells may indicate a defect in cytolytic potential of this subset during HIV-1 infection. CD56negative NK cell subsets have previously been shown to exhibit lower degranulation and IFN-γ secretion, and they were also unable to mediate antibody-dependent cellular cytotoxicity, but had similar proliferation capacities to CD56dim NK cells. 38,39,41 Indeed, assessment of the overall ability of NK cells to degranulate after overnight stimulation with K562 target cells demonstrated that the observed HIV-induced phenotype was accompanied by reduced functional activity on the CD56negative NK cell subset. Therefore, the expansion of the “pathologic” CD56negative NK cell subset accompanied by receptor alterations during chronic stimulation mediated by viral infection alters the normal functions of NK cells and may represent one of the mechanisms by which HIV-1 evades NK cell antiviral functions.
Our study was limited by a small sample size; thus, we were not able to detect the impact of the phenotypic differences on markers of disease progression. We noted significant differences in HIV RNA viral loads between the HIV-treated and HIV-viremic groups (p < .0001, Mann–Whitney, data not shown). However, no significant differences were observed between these two groups when absolute CD4 counts were compared. The timing of infection for the chronic cohort studied here could not be established, thus assessment of time since infection and association with markers of disease progression could not be determined. NK cell measurements in individuals who were treated were done after at least 12 months of initiating ART, and no associations were noted between CD56negative NK cells and CD4 recovery or viral reduction since initiation of ART for the studied group, at least in that treatment duration. Also, unavailability of longitudinal samples pre- and post-infection and after ART did not allow us to assess the overall impact over time or mechanisms behind these alterations. Given the minimal phenotypic differences observed between the treated group studied here and the viremic HIV-infected individuals, we subsequently focused only on the healthy donors and viremic HIV-infected participants for more emphasis after HIV infection. No significant differences were observed between treated and viremic HIV-infected individuals on functional assays (data not shown).
Nonetheless, our findings support findings that HIV-1 infection induces atypical phenotype on NK cells, and these abnormalities may account for the overall NK cell dysregulation and pathogenesis in chronic clade C HIV-1 infection. A better detailed understanding on the mechanisms behind this irregular phenotype is required. Alternative therapeutic strategies, beyond administration of ART, to reverse these defects and restore crucial NK cell antiviral functions necessitate future investigation.
Footnotes
Acknowledgments
The authors wish to acknowledge the Sinikithemba and Masibambisane participants from Durban, KwaZulu-Natal. The study was funded by the Doris Duke Charitable Foundation, National Research Foundation (NRF), and the University of KwaZulu-Natal College of Health Sciences, School of Laboratory Medicine and Medical Sciences.
Author Disclosure Statement
No competing financial interests exist.