
Abstract
In China, CRF01_AE and CRF07_BC are the most circulating strains of HIV-1, more and more second-generation recombinant forms have been isolated around the two strains. The same situation happened in Beijing in recent years. In our study, we have isolated a new second-generation recombinant form of HIV-1 from a male patient who was infected by homosexual behavior. Polygenetic analysis revealed that the sequence includes five CRF01_AE fragments and four CRF07_BC fragments. CRF01_AE is the skeleton of the recombinant which was inserted with four CRF07_BC fragments. The emergency of such second-generation recombinant forms manifests the diversity of the HIV-1 epidemic. Consequently, further investigation of molecular epidemiology is needed to track the genetic evolution of HIV-1.
A
The main pathogen causing the global AIDS epidemic is HIV-1; through the phylogenetic analyses of HIV-1, it could be classified into four distinct groups (M, N, O, and P), in which group M of HIV-1 is widely popular in the world. More specifically, this group of HIV-1 can be stratified into at least nine subtypes (A,B,C,D,F,G,H,J and K), five sub-subtypes (A1, A2, A3, F1, and F2), various circulating recombinant forms (CRFs), and unique recombinant forms (URFs).
4
The high rates of recombination and mutation cause the high genetic variability and extensive gene diversity of HIV-1.
5
To date, 88 CRFs have been published in the Los Alamos HIV database (
In this study, the new URF consists of 01_AE and 07_BC. The sample YA1996 was gathered in October 2015 from one male patient who was detected positive for HIV-1 in Youan hospital, Beijing. Through the epidemiological data, we realized that the patient's transmission of HIV-1 was by MSM. First, the Viral RNA was isolated from 280 μl plasma by using QIAamp Viral Mini Kit (QIAGEN, Hilden, Germany) according to manufacturer's protocol. The cDNA was generated by reverse transcription from viral RNA with Superscript III (Invitrogen, Carlsbad, CA), following the standard operating procedure. The viral near full-length genome (NFLG) was divided into two halves to amplify the overlapping regions using the same nested polymerase chain reaction (PCR) through TaKaRa LA Taq (TaKaRa, Dalian, China). All procedures were carried out in a clean room at the appropriate temperature, instruments, and specific pipettors. The products of positive PCR were purified and sequenced by SinoGenoMax (Beijing, China) with 28 specific primers. All overlapping DNA fragments were assembled and a length of 8,973 bp was gained by using Sequencher v5.1 (Gene Codes Corporation, Ann Arbor, MI).
According to the Los Alamos HIV Sequence Database (
The NFLG sequence of YA1996 in this study is 8,973 nt in length (HXB2:613-9604), spanning the gag, pol, vif, vpr, tat, rev, vpu, env, nef, and a part of 3′ long terminal repeat. The phylogenetic trees were constructed with full genome reference sequences, in which YA1996 clustered with CRF01_AE reference sequences and other 01_AE sequences with a high bootstrap value (100%). Whereas the branch of YA1996 independently existed, which indicated the presence of the recombinant (Fig. 1)
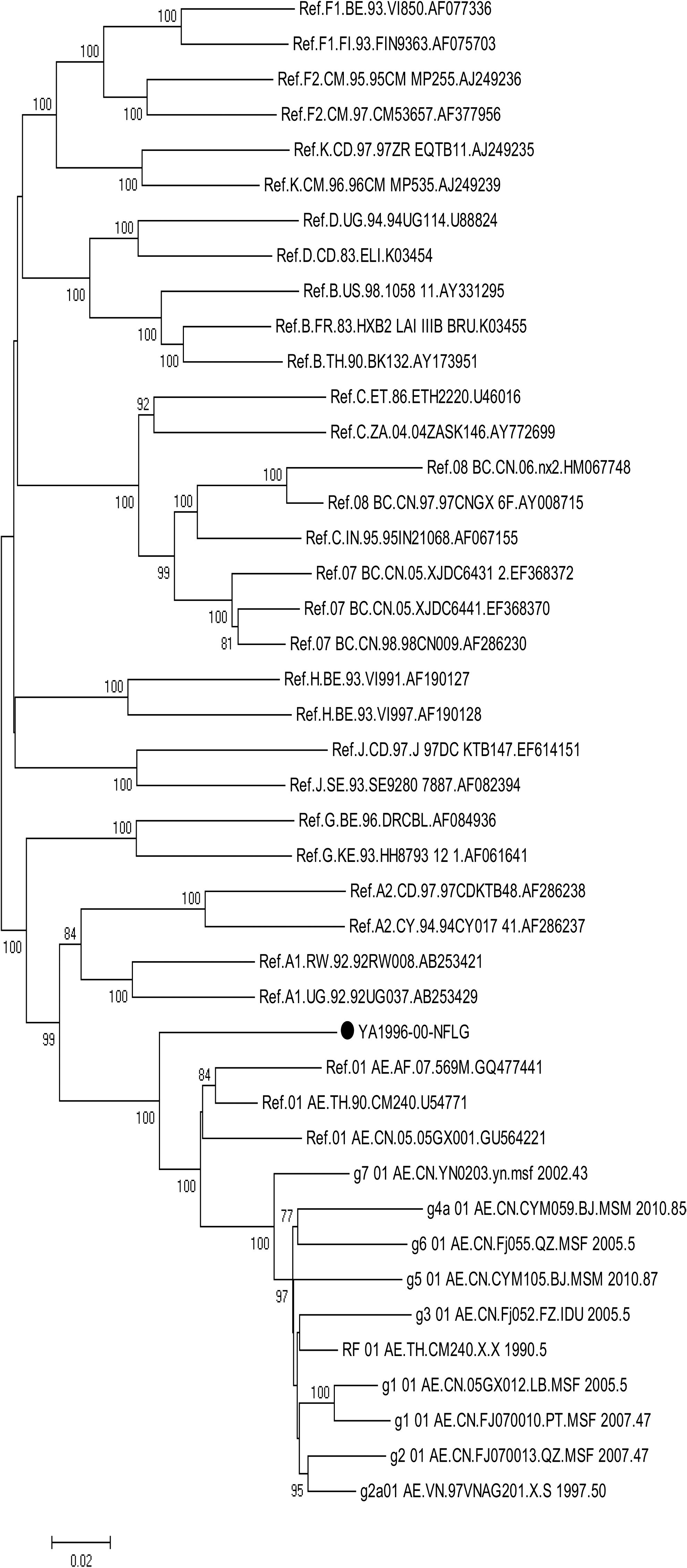
Phylogenetic tree of NFLG of YA1996 (•). The Neighbor-Joining (N-J) tree was constructed using MAGA7.0.21 based on the reference sequences and some important CRFs including CRF01_AE, CRF07_BC and CRF08_BC. All reference sequences were downloaded from the Los Alamos National Laboratory HIV sequence database (
According to the result shown in Figure 1, the sequence of YA1996 was submitted to the RIP to first identify its structure using all default settings except the window size of 300. The result manifested that the recombinant form may consist of subtypes B, C, CRF01_AE, CRF_07BC, and CRF08_BC mostly (Fig. 2A), and the particular analysis was performed by Simplot v3.5.1, indicating that the sequence most likely comprised CRF01_AE and CRF07_BC (Fig. 2B).
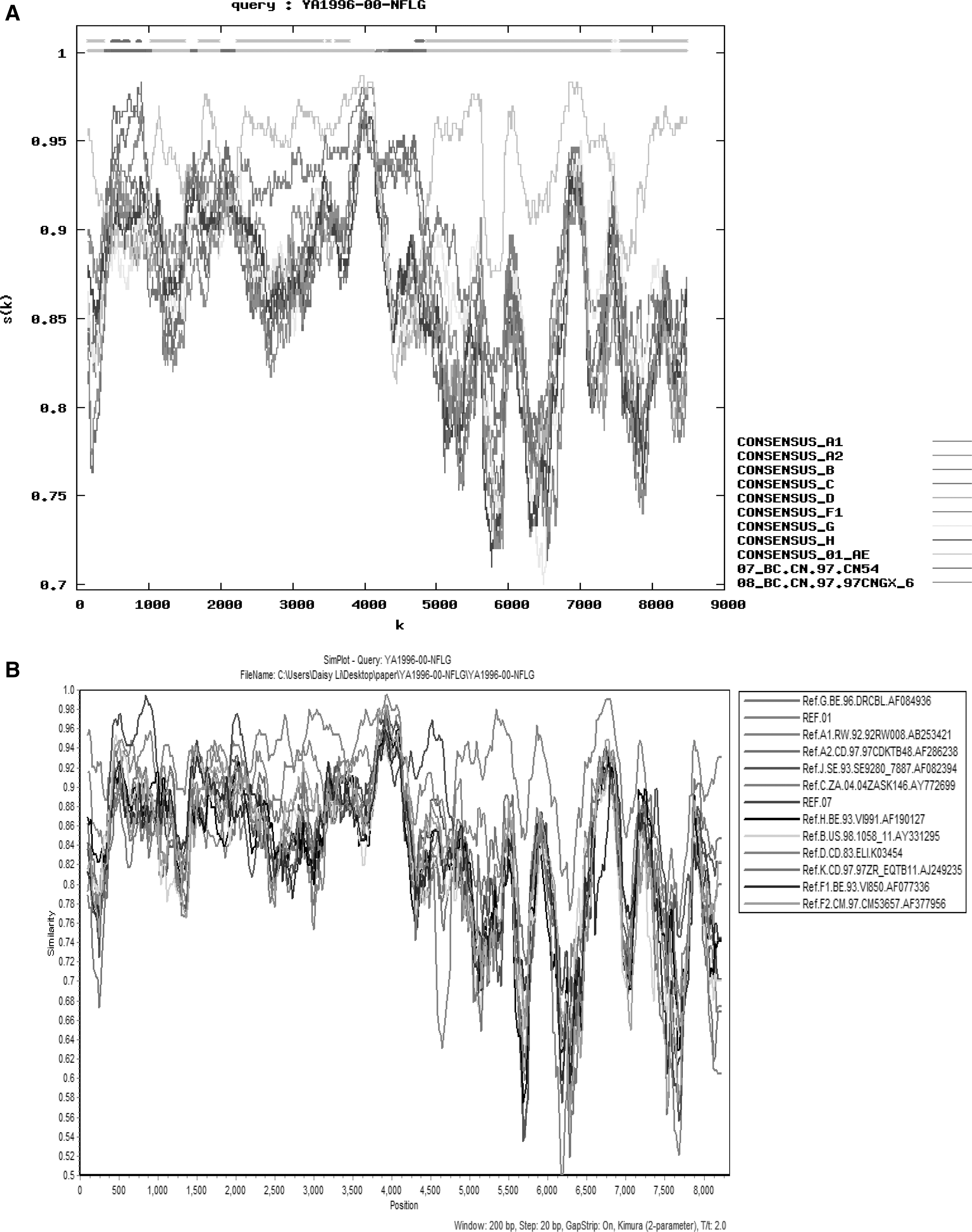
Recombinant identification program (RIP) analysis and similarity plots analysis were implemented to appraise YA1996.
Bootscan analysis was carried out to locate the breakpoint of YA1996 using Simplot v3.5.1. The query sequence of YA1996 was bootscanned with the reference sequences (CRF01_AE and CRF07_BC as parents, subtype D as outgroup). (Fig. 3A). Through jpHMM analysis, the nine unique recombination breakpoints could be identified between CRF01_AE and CRF07_BC, and location (relative to HXB2) of the sequence as follows: fragment I (790–1,169 nt) 01_AE; fragment II (1,170–1,817 nt) 07_BC; fragment III (1,818–2,801 nt) 01_AE; fragment IV (2,802–3,047 nt) 07_BC; fragment V (3,048–5,100 nt) 01_AE; fragment VI (5,101–5,674 nt) 07_BC; fragment VII (5,675–8,410 nt) 01_AE; fragment VIII (8,411–8,599 nt) 07_BC; and fragment IX (8,600–9,411 nt) 01_AE (Fig. 3B). It is obvious that the structure of the sequence is different from any known reported HIV-1 subtypes and CRFs (Fig. 3B).
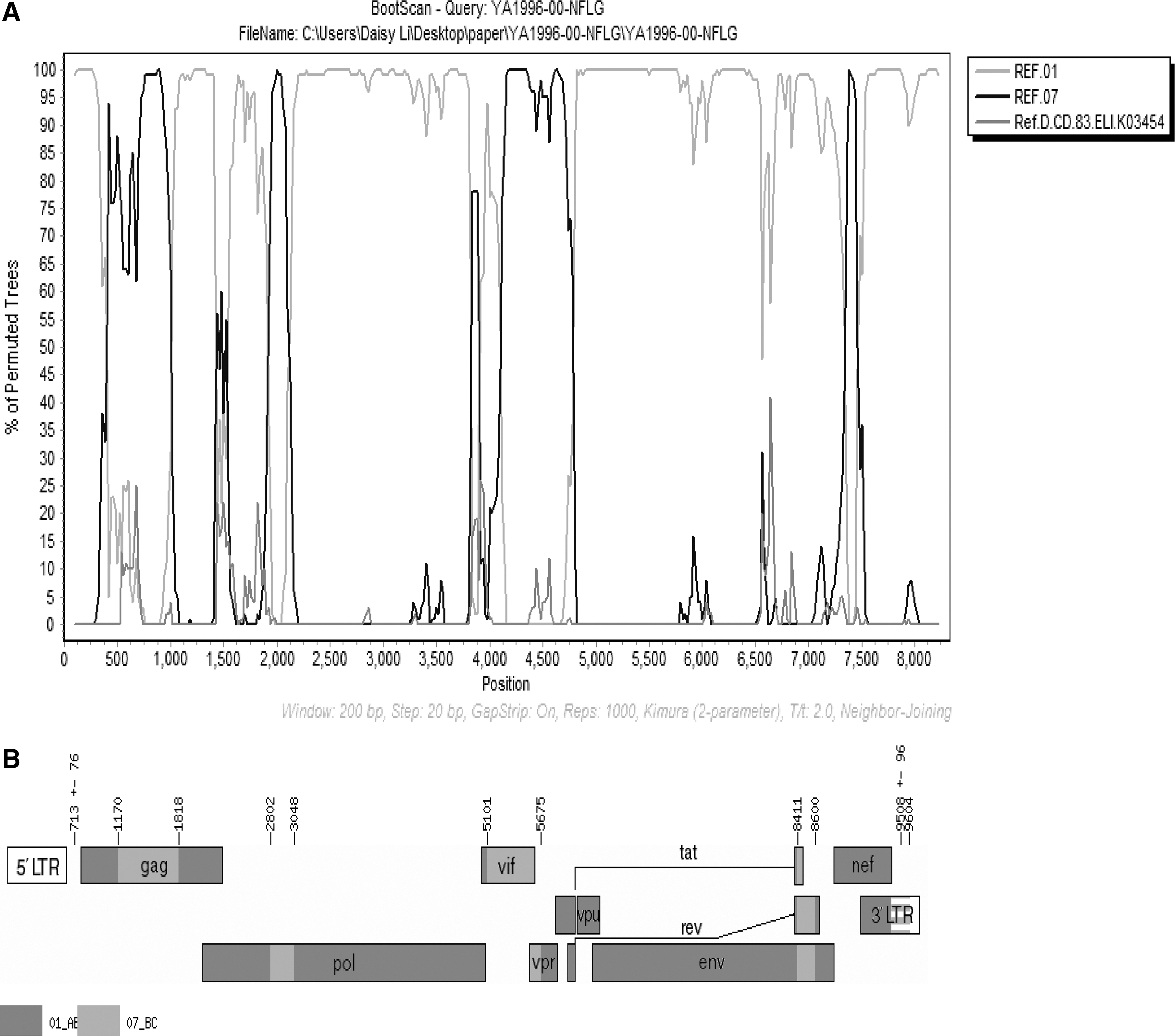
Bootscan plots and schematic representation of the YA1996 NFLG's mosaic structure.
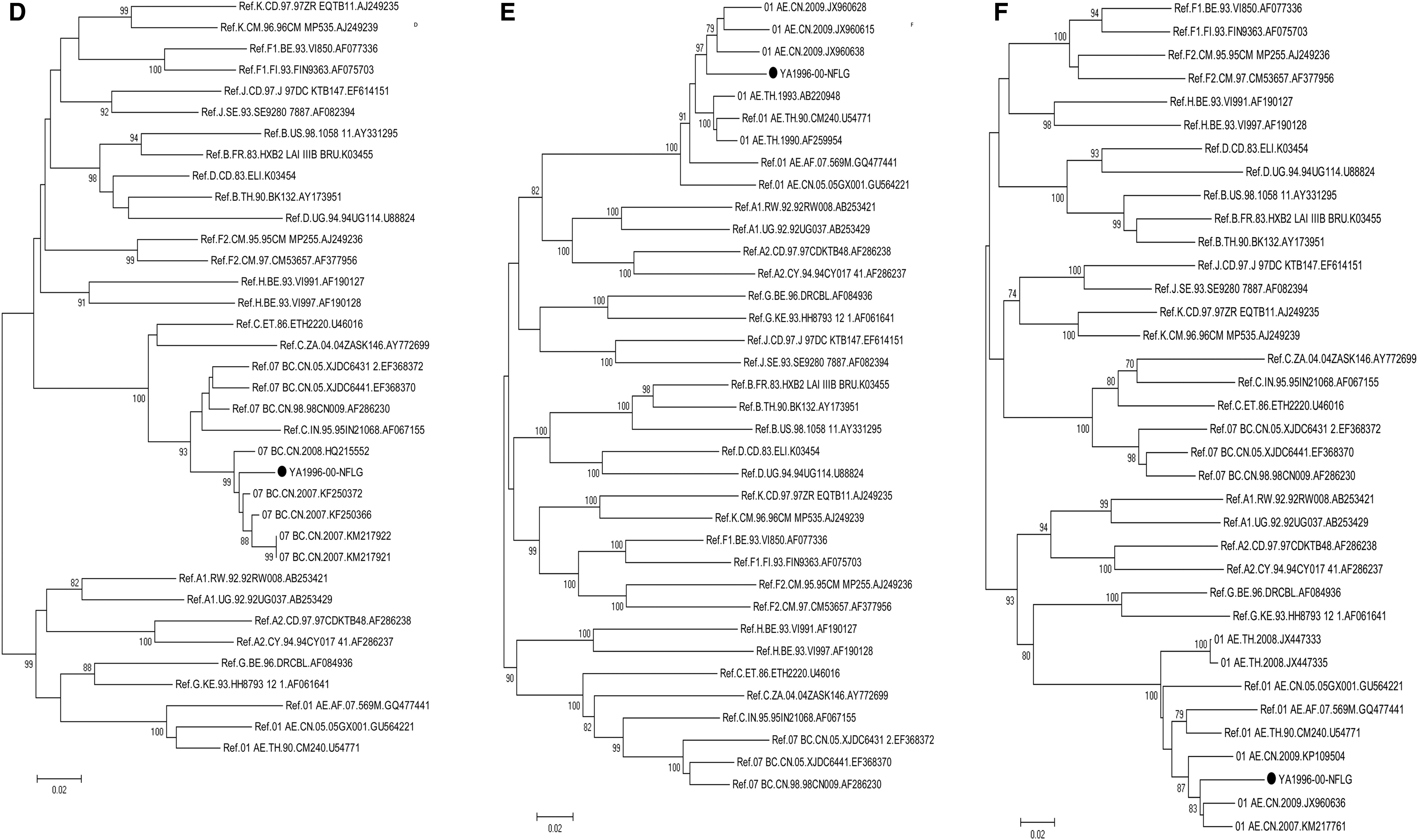
To specifically demonstrate the origin of the recombination, we have built up the subregion trees to separate each breakpoint. There were three fragments (fragments I, IV, and VIII) that were too short in size (<400 bp) to get the exact subtype among the CRFs. The analysis was performed as already mentioned, which the N-J tree was constructed with the bootstrap method by Kimura's two-parameter model. The result confirmed that the fragments II and VI were indeed clustered with CRF07_BC and fragments III, V, VII, and IX were clustered with CRF01_AE, all with a high bootstrap value (Fig. 4).

Subgenomic phylogenetic analyses of YA1996 (•), using the method described in Figure 1.
In Beijing, CRF01_AE and CRF07_BC play very important roles in the circulating strains of HIV-1, the proportion of which is 36.7% and 20.9%, respectively. 7 In China, CRF01_AE is the major strain in the heterosexual epidemic. 9 Gradually the MSM population has increased with time. The CRF07_BC was firstly isolated from intravenous drug users (IDUs) in southwest China, 10 and then it emerged in MSM population, which may indicate that there was cross-transmission between different population.
According to the report, the incidence of URFs in newly diagnosed treatment-naive patients was 4.4% (33/712) based on the analysis of pol gene, in Beijing, 2016. 11 In addition, the emergence of second-generation recombinant was 3.4% (8/237) among the students in Beijing. 12 As an international city, the flow of personnel is large in Beijing. A survey of MSM population behavior in Beijing showed that 69.8% (349/500) of the MSM population had more than two male sexual partners in the past 6 months. High-risk sexual behavior with multiple sexual partners and unprotected sex among the MSM population can easily lead to HIV recombinants in different subtypes, which contributes to the generation of new recombinant strains. 13
In the same group or in the same area, coexistence of CRF01_AE and CRF07_BC provides an opportunity to generate a new recombinant form. The initial URF of CRF01_AE and CRF07_BC was reported in Jiangsu province among IDUs, 14 after that other provinces in China have isolated it with different forms among MSM and heterosexual population. 15,16 In this study, we have identified a new URF in Beijing area, which contains different break points between CRF01_AE and CRF07_BC strains. However, the emergence of many new URFs raises difficulties in AIDS prevention and control work. In this situation, the prevalence of new recombinant forms of HIV should be paid more attention in the future.
Sequence Data
The nucleotide sequence of YA1996 has been submitted to GenBank with the accession number MF084205.
Footnotes
Acknowledgments
The authors thank the doctors and nurses at Youan hospital for their admirable treatment and patient care. This study is supported by Beijing Science and Technology Planning Project of Beijing Science and Technology Commission (D161100000416002).
Author Disclosure Statement
No competing financial interests exist.