
Abstract
The inherent stability of a small population of T cells that are latently infected with HIV despite antiretroviral therapy (ART) remains a stubborn obstacle to an HIV cure. By exploiting the memory compartment of our immune system, HIV maintains persistence in a small subset of quiescent cells with varying phenotypes, thus evading immune surveillance and clinical detection. Understanding the molecular and immunological mechanisms that maintain the latent reservoir will be critical to the success of HIV eradication strategies. Human cytomegalovirus (CMV), another chronic viral infection, frequently co-occurs with HIV and occupies an oversized proportion of memory T cell responses. CMV and HIV have both evolved complex strategies to manipulate our immune system for their own advantage. Given the increasingly clear links between CMV replication, chronic immune activation, and increased HIV reservoirs, we present a closer examination of the interplay between these two chronic coinfections. Here we review the effects of CMV on the immune system and show how they may affect persistence of the latent HIV reservoir during ART. The studies described herein suggest that hijacking of cytokine and chemokine signaling, manipulation of cell development pathways, and transactivation of HIV expression by CMV might be pouring gas on the fire of HIV persistence. Future interventional studies are required to formally determine the extent to which CMV is causally associated with inflammation and HIV reservoir expansion.
Introduction
I
Three main mechanisms contribute to HIV persistence during suppressive ART: first, the inherent stability and longevity of a small population of resting T cells that are latently infected with HIV despite long-term suppressive ART. 5 Second, latently infected cells can proliferate in response to antigenic stimulation or cytokines. 6 Third, HIV can be expressed at low levels despite ART, especially in certain immunological sanctuaries, promoting immune activation and further enhancing the recruitment of target cells. 3
The presence of CMV coinfection might add a new dimension to each of these putative mechanisms of HIV persistence. CMV has a high worldwide prevalence with estimates ranging from 40% to 100% seropositivity, and establishes a life-long infection characterized by persistent antigen exposure and interaction with the host immune system. CMV prevalence is further concentrated among older people and people living with HIV (seroprevalence of 80%–100%). 7 –9 In general, CMV is only pathogenic in individuals with deeply compromised immune systems, but people living with HIV often have asymptomatic, ongoing CMV replication in various mucosal sites and effector tissues. 10 For example, we recently showed that the vast majority of HIV-infected men who have sex with men (MSM) had evidence of persistent CMV replication in their genital secretions at one or multiple time points throughout 1 year of follow-up. 11
This subclinical CMV replication has been associated with T cell dysfunction and with impaired immune recovery after HIV infection. 12,13,14 As we discuss in this review, persistent CMV replication can also promote longevity and proliferation of HIV-infected cells, the recruitment of new HIV target cells, and directly enhance HIV transcription. Taken together, we hypothesize that coinfection with CMV could be an important factor contributing to the establishment and maintenance of the HIV reservoir during ART.
CMV Coinfection and HIV Persistence
Recent studies suggest that coinfections such as CMV can exacerbate HIV-related chronic immune activation during ART, and might augment the size of the latent HIV reservoir. 1,2,15 For example, a longitudinal study following 107 men starting ART during the earliest phase of their HIV infections (median 3 months since estimated date of infection) over a median follow-up period of 19 months observed an association between presence of CMV and Epstein–Barr virus (EBV) replication in peripheral blood mononuclear cells and slower decay rate of the HIV DNA reservoirs during ART. 1 Other cross-sectional studies demonstrated that subclinical CMV replication is associated with higher levels of HIV DNA in both ART-naive and ART-suppressed individuals. 2,16 Interestingly, in a small cohort of people living with HIV who underwent myeloablative chemotherapy, HIV DNA was enriched in CMV- and EBV-specific CD4+ T cells after immune reconstitution. 15 Although we cannot infer causality due to the observational design of these studies, these findings support a model in which CMV replication, increased HIV reservoirs, and inflammation are inextricably linked.
A variety of possible concurrent mechanisms may account for the relationship between CMV and HIV persistence. Specifically, CMV may promote HIV persistence through (1) altered chemokine receptor-mediated cell trafficking to sites of inflammation, (2) induction of inhibitory immune pathways (e.g., cellular exhaustion, PD-1, and IL-10 expression), (3) proliferation and clonal expansion of HIV-target cells, (4) inhibition of apoptosis, and/or (5) direct transactivation of latent HIV (Fig. 1). Here, we review what is known about these mechanisms in the context of HIV and CMV coinfection.
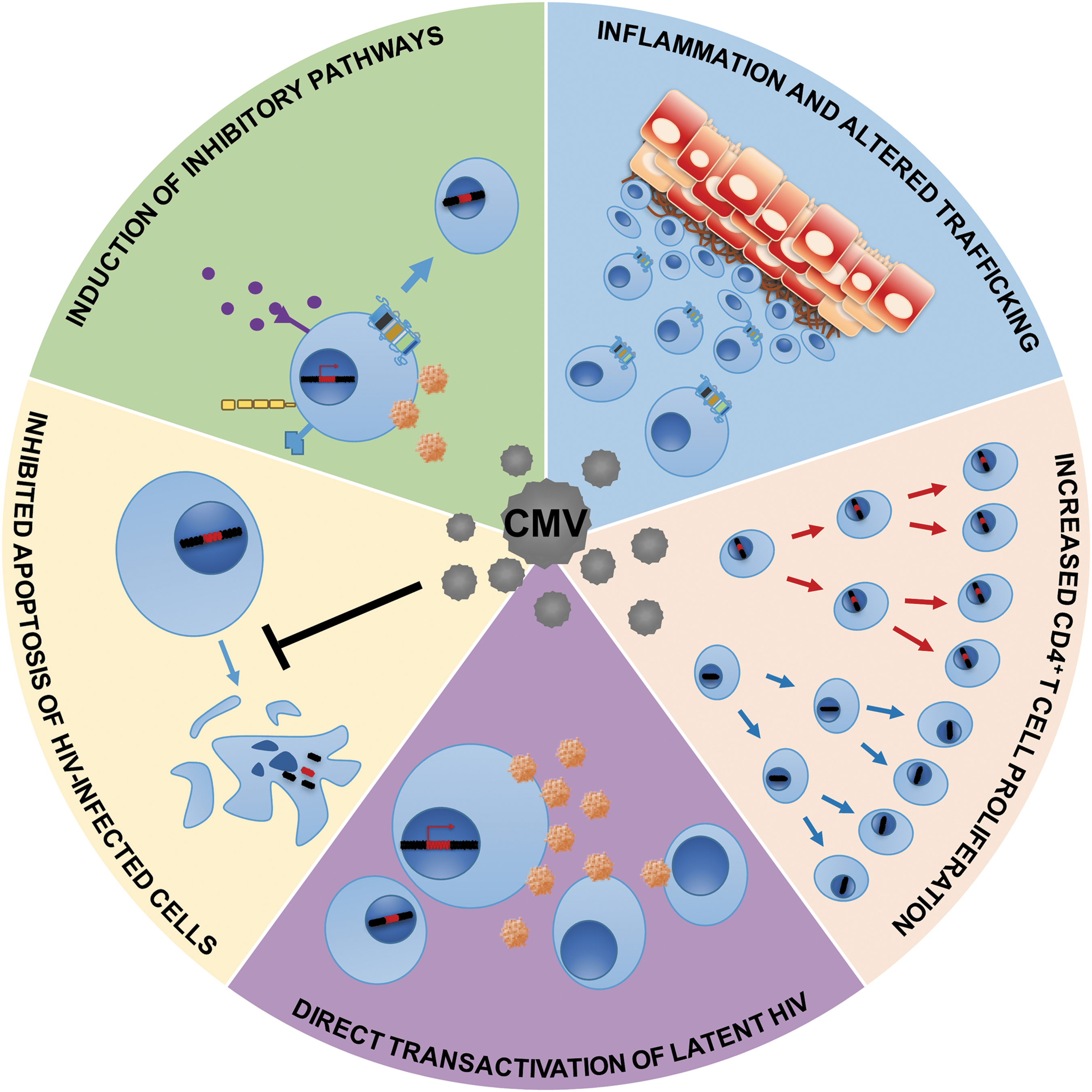
Putative mechanisms by which CMV might promote HIV persistence. Cells harboring latent HIV could become more numerous through CMV-associated inflammation, inhibitory signaling, proliferation, or inhibition of apoptosis. CMV can also directly transactivate latent HIV, which could lead to more target cells becoming infected. Blue circles with nucleus represent CD4 T cells with and without integrated HIV DNA; purple dots represent inhibitory cytokines; orange circles represent HIV. Gray circles represent CMV. Squared cells represent epithelial cells with signs of inflammation. CMV, cytomegalovirus.
Inflammation and altered chemokine trafficking
In a complex host–virus relationship, CMV elicits and maintains a high frequency of virus-specific T cells that engage in a life-long effort to restrain CMV replication and prevent disease. 8,17 Since CMV replication is enhanced by inflammatory stimuli, the virus developed ingenious strategies to induce and augment inflammation. 18 For example, CMV directly upregulates the expression of several cytokines and inflammatory mediators in host cells, including IL-1β, IL-6, and type I interferon, triggering inflammatory responses. 19 –22 CMV and other herpesviruses can also upregulate IL-15, 23 which is a common γ-chain cytokine like IL-2 and IL-7. This group of cytokines can promote the expansion of CD4+ and CD8+ T cells independently of antigen specificity, making them particularly relevant to the HIV reservoir. 23 –25
CMV encodes its own cytokines and chemokine homologues as well as cytokine receptor homologues that can further modulate levels of human cytokines, chemokines, and growth factors. 19,26,27 For example, CMV alters the expression of the chemokines RANTES, MCP-1a, and IL-8, 28 and at least six CMV genes (UL33, UL78, UL146, UL147, US27, and US28) encode chemokine-receptor-like protein. Furthermore, CMV-exposed central memory T cells from cord blood mononuclear cells demonstrated increased expression of CCR5 protein, 29 which is the main coreceptor for HIV cellular entry. 30 Taken together, the CMV-induced proinflammatory milieu and disrupted chemokine trafficking could promote the influx of HIV target cells at sites of HIV persistence.
Consistent with our hypothesis that CMV increases HIV persistence by recruiting HIV target cells to sites of inflammation, a recent study of ART-treated people living with HIV demonstrated that CMV replication in the gut was associated with disrupted mucosal barrier integrity, microbial translocation, and inflammation. 3 CMV-induced inflammation in the gut—a major site of HIV replication—likely attracts HIV target cells or increases the susceptibility of the local immune milieu to HIV infection. 3,31 The specific effects of CMV replication and CMV-induced inflammation on the trafficking of HIV target cells remain poorly defined in humans, but merit deeper investigation. The hijacking of cytokine and chemokine signaling by CMV leads to an inflammatory environment that is ripe with HIV target cells.
Induction of inhibitory immune pathways
Not only does CMV induce inflammation but it can also hinder immunity directed toward CMV-infected cells, 32,33 which may have important implications for HIV persistence. Like HIV, CMV can inhibit human leukocyte antigen (HLA) expression on cellular surfaces, thereby impairing antigen presentation. 34 However, compared with HIV, the larger genome of CMV allows for an expanded arsenal of immune modulatory factors. 28,32,34 For example, CMV can produce decoy viral homologues of HLA class I molecules that allow it to avoid immunosurveillance by natural killer (NK) cells. 32 Such immune diversions—developed by CMV over millions of years of coexistence with the human species—could favor immune evasion not only for CMV itself but also for other coinfecting viruses, such as HIV.
CMV has also evolved strategies to hijack cell-signaling pathways, such as PD-1 and IL-10, and the disrupted signaling may coincidentally promote HIV persistence. 14,26,27,33,35,36 For example, in a recent study of 45 MSM who were virally suppressed on ART, seminal CMV shedding was associated with increased PD-1 expression on CD4+ T cells. 14 This may be important because HIV-specific T cells are enriched in PD-1 expression, and PD-1 expression on CD4+ T cells is positively correlated with HIV viremia. 37,38 Induction of PD-1 signaling on CD4+ T cells has been postulated as a mechanism to enable recently infected CD4+ T cells to shift toward a state of persistence, rather than one of activation-induced cell death. 39,40 PD-1 expressing CD4+ T cells are highly enriched with integrated HIV DNA, 39 and expression of PD-1 by CD8+ T cells causes impaired HIV-specific immunity. 41,42
IL-10 is a cytokine that promotes tolerance and potently stifles the proinflammatory activities of many immune cell types by (1) promoting the development of regulatory T cells, (2) diminishing the proliferative capacity of lymphocytes, (3) inhibiting the ability of monocytes to activate T cells in response to antigen, (4) skewing the cytokine expression of monocytes, and (5) blunting the effector functions of lymphocytes (reviewed in Ref. 43 ). HIV itself can stimulate the production of IL-10 in vitro, 44 and IL-10 can inhibit latency reversal in CD4+ T cells and primary human macrophages in vitro. 45 This theory is supported by lymphocytic choriomeningitis virus models, which show that upregulation of IL-10 production by antigen-presenting cells is associated with impairment of T cell functions, thus allowing viral persistence. 46,47 Inhibition of HIV replication by IL-10 has been demonstrated in vitro, 48 and high IL-10 plasma levels are associated with the control of viral replication during pregnancy. 49 This has implications for the reservoir because otherwise productively infected cells may become quiescent through IL-10 expression.
Interestingly, CMV encodes for several viral homologues of human IL-10 (hIL-10) produced through alternative splicing of the UL111A gene. 50 The predominant form of viral IL-10, cmvIL-10, which despite having only 27% amino acid sequence identity to hIL-10, binds with high affinity to the hIL-10 receptor. 51 Furthermore, cmvIL-10 triggers the same anti-inflammatory effects as hIL-10. In particular, cmvIL-10 impairs maturation of dendritic cells, downregulates surface HLA expression, inhibits inflammatory cytokine production and cell proliferation, activates transcription factor Stat3, and upregulates hIL-10 expression to further promote an immunosuppressive environment during infection (reviewed in Ref. 52 ). The second predominant viral IL-10, latency-associated cmvIL-10 (LAcmvIL-10), has been described in a model of latently infected granulocyte macrophage progenitor cells. 53 LAcmvIL-10 lacks some IL-10R contact residues because of its truncated C-terminus and thus retains some, but not all, of the immunosuppressive functions of cmvIL-10. 53 CMV-mediated IL-10 signaling participates in preventing immune responses, thus indirectly favoring HIV persistence. Collectively, these findings suggest that HIV and CMV both hijack IL-10 and PD-1 signaling. Future studies should address the mechanisms by which IL-10 and PD-1 benefit CMV and HIV replication, or if they influence the size of the latent reservoir in vivo.
Proliferation and clonal expansion of HIV-infected cells
The homeostatic proliferation of memory T cells is one mechanism by which we maintain long-lived immunological memory. Stem cell memory T (Tscm) cells are particularly relevant to this process and to the HIV reservoir, as Tscm cells from people with HIV have the highest levels of per cell copies of integrated provirus. 54 Homeostasis of the latent HIV reservoir can be preserved, in part, through the self-renewal capacity of the Tscm cell subset. 54 Functional, CMV-specific Tscm cells are present in people with CMV, but absent in CMV negative control donors. 55 Therefore, it is theoretically possible for a subset of CMV-specific stem cells to replenish the HIV reservoir despite ART through homeostatic proliferation.
Evidence that clonal expansion contributes to HIV persistence is even more compelling. Genetic analyses of HIV proviruses and integration sites in people with HIV who are ART-suppressed provide strong evidence for clonal expansion of HIV-infected CD4+ T cells as a crucial mechanism of viral persistence during suppressive ART. 56 –60 Self-renewal capacity of memory CD4+ T cells is important to maintain immunological memory and can be driven by homeostatic responses to CD4+ T cell depletion and inflammation, or directly by antigen stimulation (such as HIV or CMV). 61,62 Antigen dose affects immune responses, potentially turning initial inflammation into immune exhaustion at different rates. 63 CMV antigens in particular are chronically present at high doses, 64 and consequently CMV-specific T cells account for a disproportionately large subset of total T cell memory responses. 17 This proportion is even greater among HIV-infected adults, and remains high after ART-mediated suppression of HIV replication. 65,66 Although CMV-specific T cells are relatively resistant to de novo HIV infection, 67 CMV-associated immune activation could nonetheless induce proliferation of T cells, including HIV-infected cells. If CMV-induced T cell proliferation preferentially affects HIV-infected cells, this may lead to an increase in the HIV latent reservoir.
Studies involving self-renewal and persistent antigenic stimulation of CD4+ T cells leave many unanswered questions. For instance, we do not know whether HIV provirus can compartmentalize and concentrate in distinct subsets of the adaptive immune system, such as CMV-specific cells. If the latent HIV reservoir continuously re-establishes itself in response to CMV-derived antigenic stimulation of the immune system, one might expect that CMV-specific cells would demonstrate heightened levels of proviral DNA with evidence of evolution over time, but this has not been investigated to our knowledge. Little is known about the effects of subclinical CMV replication on key T cell subsets that contribute to persistence, such as Tscm cells, central memory T cells, transitional memory T cells, and CD4+ T cells expressing PD-1, TIGIT, and LAG3. 41,68 IL-7- and IL-15-mediated expansion of HIV-infected cells also contribute to persistence during virally suppressive ART, 69,70 and the influence of CMV on this process is also unknown.
Inhibition of apoptosis
Abortive HIV infections can induce cell death through cellular sensors of viral products. 71,72 The rapid loss of CD4+ T cells during acute HIV infection is due, in part, to the innate cellular defenses that can trigger apoptosis before HIV successfully integrates. This is an especially important pathway in naive T cells, which exhibit heightened antiviral defenses but are prone to remain quiescent and latent upon successful HIV integration. 73 CMV may interfere with protective immunity against HIV through the blocking of this defensive apoptosis: CMV is armed with two proteins that inhibit apoptosis, viral mitochondrial-localized inhibitor of apoptosis (vMIA) and viral inhibitor of Caspase-8-induced apoptosis (vICA). 74 vMIA blocks apoptosis by inhibiting cytochrome C release from mitochondrial membranes and vICA blocks apoptosis by interfering with the activation of Caspase 8 (reviewed in Ref. 75 ). Neither of these proteins has been studied specifically in the context of HIV-infected cells. Blocked apoptosis might, therefore, allow a more robust seeding of the latent reservoir, especially during acute HIV infection. CMV-encoded inhibitors of apoptosis should be evaluated for their effects on the size of the HIV reservoir and their ability to promote de novo HIV infections of quiescent cells.
Direct transactivation of latent HIV by CMV
Although CMV does not generally infect CD4+ T cells, CMV replication could nonetheless promote inflammation and HIV persistence by inducing compartmentalized, ongoing HIV RNA expression in effector tissues, despite ART suppression. This is likely made possible through secreted mediators of inflammation and/or altered cell-mediated immunity, since HIV preferentially integrates into actively transcribed regions, rendering its expression particularly susceptible to regulation by host transcription factors. 76 Evidence from as early as 1990 has supported the concept that CMV (especially immediate early genes) can induce HIV expression by manipulating cellular transcription pathways. 77 In a related study, CMV infection significantly increased HIV long terminal repeat (LTR)-driven reporter gene expression in transfected human fetal astrocytes. 78
Activation of mammalian target of rapamycin (mTOR) signaling may be involved in the induction of HIV expression by CMV as well. 79,80 mTOR activation is a common tactic employed by intracellular pathogens to benefit their own replication, 81 and both CMV and HIV activate mTOR signaling. 79,80 Moreover, inhibition of mTOR signaling was shown to not only impair HIV entry and transcription but also decreases viremia in a humanized mouse model of HIV infection. 82 Interestingly, mTOR also regulates proliferation and inflammation pathways (reviewed in Ref. 81 ), underscoring the potential importance of mTOR in enabling the chronic, pathogenic effects of HIV and CMV coinfection. In a human monocyte cell line, CMV was shown to induce activation of AP-1 and NF-kappaB. 83 These transcription factors are critical for efficient transcription of integrated HIV, and NF-kappaB has also been implicated in the establishment of latency. 84
The effects of CMV on the transcriptional environment of HIV-infected cells are complex, but few studies directly address this topic in vivo, despite the high prevalence of CMV coinfection in people living with HIV. Transacting factors that mediate CMV-induced transcriptional dysregulation, and its effects on latency or HIV expression, merit further investigation.
Conclusions
The latent reservoir is not static, and the mechanisms by which the latent HIV reservoir is replenished are still poorly understood. Defenses evolved by CMV to commandeer the immune system might create a favorable environment for HIV reservoir seeding, thus representing an additional hurdle to be addressed by successful HIV eradication strategies.
Multiple mechanisms may be responsible for larger HIV reservoirs in the presence of CMV replication (Fig. 1). By changing the development, trafficking, proliferation, apoptosis, and/or transcriptional environment of HIV target cells, CMV slows the decay of the HIV reservoir during ART. 1 The degree to which each mechanism contributes to the expansion of the HIV reservoir in vivo—under which circumstances—remains unclear, but it is likely that many of them occur simultaneously. For example, persistent inflammation due to CMV could also affect proliferation, susceptibility to apoptosis, and latent HIV transactivation. Thus, synergistic effects between these mechanisms should also be considered. One additional caveat to many CMV and HIV studies is that increased CMV replication could be a consequence of increased inflammation or larger HIV reservoirs. Interventional studies will be required to determine the degree and direction of causality between CMV, HIV, and inflammation.
Many of the strategies aimed at curing HIV use pharmacological or biological agents to stimulate cells harboring latent HIV, to purge the latent reservoir. 85 However, activation of latent HIV DNA with immune modulatory interventions could affect replication of CMV and other human herpesviruses, which might, in turn, limit HIV clearance. The effects of latency reversing agents and immune modulatory therapies on CMV reactivation are currently unknown and should be evaluated in clinical trials. Future studies should assess the replication competence of HIV DNA reservoirs in the setting of CMV replication, and should seek to determine whether CMV-induced cell survival and proliferation preferentially affect HIV-infected cells. By examining the role of CMV in promoting HIV persistence, we may reveal pathways to disrupt the latent HIV reservoir, thus aiding successful cure strategies.
Footnotes
Acknowledgments
We are grateful to Antoine Chaillon for his help designing our figure. We also appreciate Lisa Loeb Stanga and the California HIV Research Program for constructive feedback. This work was supported primarily by a grant from the National Institutes of Health, University of California, San Francisco-Gladstone Institute of Virology & Immunology Center for AIDS Research, P30-AI027763 (CNIHR), California HIV Research Program Ideal award to Sara Gianella, by the department of Veterans Affairs, the James B. Pendleton Charitable Trust and additional grants from the National Institutes of Health: AI100665, MH100974, MH097520, DA034978, AI007384, AI027763, AI106039, AI43638, AI074621, AI036214, MH101012, UL1TR000100, CARE U19 AI096113, and AI068636-09. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Authors' Contributions
A.C.-Q., C.V., A.L., and S.G. participated in creating the outline design, literature search, and wrote the primary version of the article. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.