
Abstract
Cytotoxic T cells are critical in controlling virus infections. However, continuous antigen stimulation and negative regulatory factors cause CD8 T cells to enter a dysfunctional state (T cell exhaustion), resulting in viral persistence. We hypothesized that the exhausted T cell state could be molecularly rejuvenated using a somatic cell reprogramming technology, which is technically able to convert any types of cells to induced pluripotent stem cells (iPSCs), to regenerate functional T cells capable of purging chronic infection. We generated a new mouse line (B6/129OKSM) in which every somatic cell contains four doxycycline-inducible reprogramming genes (Oct4, Klf4, Sox2, and c-Myc: OKSM), and infected them with lymphocytic choriomeningitis virus (LCMV) clone 13 to establish chronic infection. Exhausted LCMV-specific T cells isolated by flow sorting were successfully reprogrammed ex vivo into iPSCs in the presence of doxycycline. Upon injection into blastocysts and subsequent transfer into foster females, the reprogrammed cells differentiated into functional naive T cells that maintained their original antigen specificity. These results provide proof of concept that somatic cell reprogramming of exhausted T cells into iPSCs can erase imprints of their previous exhausted state and in turn regenerate functional virus-specific T cells.
Introduction
T
T cell exhaustion is a long-term dysfunctional state caused by extensive cellular programming 1,2 and was originally identified in mice infected with lymphocytic choriomeningitis virus (LCMV). 3 –5 Exhausted T cells have also been observed in response to other virus infections, such as simian immunodeficiency virus (SIV), human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV). 6 –13 These cells can be characterized by a significant decrease in cytokine production, such as IL-2, IFN-γ, and TNF-α, as well as by cell cycle arrest. 14 –16 Programmed death-1 (PD-1, also known as CD274) was the first proposed exhausted T cell marker. 17 Rescue of effector functions of exhausted T cells through disruption of PD-1 binding with its ligands (PD-L1 and PD-L2) was demonstrated in various chronically infected animal models, including LCMV-infected mice, 17 HIV-infected humanized mice, 18 and SIV-infected nonhuman primates. 19,20 However, activated T cells also express PD-1 and remain functional, 21 indicating that PD-1 is neither a sole marker for exhausted T cells nor an inhibitory surface receptor.
Therapeutic interventions exploiting the PD-1/PD-L1 interaction have been shown to enhance antiviral T cell function and lead to increased control of chronic virus infections in vivo, 19,20 demonstrating the powerful potential of T cell immunotherapy. However, since the dysfunctional cellular program remains, once therapeutics are withdrawn, T cells can again lose antiviral function. 22 Thus, novel methods to functionally reprogram these exhausted T cells to effectively and durably fight infection are needed to control these infections. Importantly, adoptive transfer of functional memory T cells effectively eradicates chronic LCMV infection in mice, and HBV and multiple herpes virus infections in humans, 23 –25 suggesting that if exhausted T cells could effectively be reprogrammed into functional memory T cells while maintaining their original antigen specificity, it may be feasible to use these cells to control the infection.
Reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) has contributed greatly to our understanding of factors responsible for cell phenotypes and realization of their potential application toward future clinical use. 26 The exhausted T cell state is recognized as an alternative, and thus far, irreversible program of T cell differentiation. 27 Yet, the permanence of the exhausted program and the ability to erase it by de-educating the cells back to a naive state are still unclear. Thus, we hypothesized that molecularly reprogramming exhausted virus-specific T cells into iPSCs and then redifferentiating them into naive T cells could erase their previous dysfunctional imprints while retaining their virus specificity.
Chronic viruses rapidly evolve within an individual. As a result, each individual's antivirus TCR repertoire is person specific. In addition, each individual has a donor-specific human leukocyte antigen (HLA) that limits current attempts to clone and engineer single TCRs 28 restricted to one HLA haplotype. Thus, a broad and nonspecific approach may not be as effective in engineering immunity to fight chronic virus infections compared with using each individual's own T cells, provided the function of exhausted T cells can be reprogrammed. Furthermore, the ability to broadly reprogram virus-specific T cells would allow for the breadth of response needed to prevent virus escape from a single reprogrammed epitope or engineered TCR. Although redifferentiated T cells from reprogrammed CTL clones had some ability to kill HIV-infected cells, 29 the functional status of the cells before reprogramming could not be ascertained in their system used in the report because of the massive ex vivo amplification and TCR stimulation during CTL clone generation. Due to the system being used, it is unclear whether the reprogrammed cells were derived from initially exhausted cells or whether the ex vivo culture restored their function before reprogramming. 30 Importantly, in those in vitro systems, it is difficult to experimentally measure (1) the extent of molecular and functional restoration compared with parental exhausted T cells, (2) the identification of functional/transcriptional/epigenetic imprints retained through reprogramming and how these affect subsequent antiviral function/development, (3) the ability to generate effective immune memory, and ultimately (4) the ability to assess the reprogrammed cells' ability to eliminate the chronic infection from which they were derived. Thus, the efficacy and molecular nature of T cell reprogramming, whether exhausted T cells retain specific imprints of their previous differentiation state that then limit antiviral activity, the ability to generate functional T cells following somatic cell reprogramming, and the utility of using reprogrammed exhausted T cells to fight chronic virus infection, are critical questions that need to be defined to begin to determine how T cell reprogramming approaches can be used to treat chronic virus infections.
Our overall goal is to provide proof of concept that exhausted virus-specific T cells from chronic infection can have their dysfunctional genetic programs erased by a somatic cell reprogramming technology, and that a clean slate for proper and effective antiviral activity as T cells with their original virus specificity for immunotherapeutic purposes can be instituted. In this study, we used mice infected with LCMV clone 13 (LCMV CL13) to test this hypothesis. This model system has been well recognized as a murine model for establishing chronic viral infection with thorough characterization of T cell responses that parallel those of HIV, HBV, and HCV infections. 12,31,32 Thus, our approach for restoring functional antiviral activity from exhausted T cells could be applicable for other chronic infections.
Materials and Methods
Cells and reagents
AinV15 mouse embryonic stem cells (ESCs) were purchased from American Type Culture Collection (ATCC SCRC-1029, Manassas, VA). Mouse ESCs and iPSCs were maintained in ESC medium [15% FCS (SH30070.03E; GE Healthcare Life Sciences, Marlborough, MA) in Knockout DMEM (10829018; Thermo Fisher Scientific, Canoga Park, CA), with 1% GlutaMAX (Thermo Fisher Scientific), 1% MEM nonessential amino acid (Thermo Fisher Scientific), 1,000 U/ml leukemia inhibitory factor (ESG1106; Millipore Sigma, St. Louis, MO), and 0.1 mM 2-mercaptoethanol (EmbryoMax; Millipore Sigma)]. Doxycycline (DOX) was also obtained from Millipore Sigma (D3072). γ-irradiated mouse embryonic fibroblasts (iMEF) derived from CF-1 mouse line were purchased from MTI-GlobalStem (GSC-6301G, Gaithersburg, MD). Embryoid bodies were generated as reported. 33 Antibodies were purchased from the following sources: SSEA-1 (MC-480; BioLegend, San Diego, CA), Nanog (M55-312; BD Biosciences, San Jose, CA), Fibroblast marker (ER-TR7; Santa Cruz Biotechnology, Dallas, TX), mouse CD19 (1D3; Thermo Fisher), mouse/human B220 (RA3-6B2, BioLegend), mouse CD3 (17A2; BioLegend), mouse CD4 (RM4-5; BioLegend), mouse CD8 (53-6.7; BioLegend), mouse CD62 L (MEL-14; BioLegend), mouse CD25 (PC61; BioLegend), mouse PD-1 (29F.1A12; BioLegend), and CD44 (IM7; BioLegend). Matrigel qualified for human ESC culture was purchased from BD Biosciences. Magnetic beads coated with anti-CD3 and anti-CD28 antibodies were purchased from Thermo Fisher (Dynabeads mouse T-activator CD3/CD28, Thermo Fisher Scientific). LCMV gp33–41 (KAVYNFATM) tetramer and LCMV gp276-286 (SYVENPGGYCL) tetramer, both of which are mouse H-2Db restricted, were generated by the NIH Tetramer Core Facility at Emory University. The latter was used as a negative control.
The primers were purchased from IDT (Coralville, IA; Table 1).
Animals
Animal research described in the study was conducted under UCLA's Chancellor's Animal Research Committee (Institutional Animal Care and Use Committee [IACUC]) in accordance with guidelines for housing and care of laboratory animals of the National Institutes of Health (NIH) and the Association for the Assessment and Accreditation of Laboratory Animal Care (AALAC) International. Chimeric mice from iPSCs were generated at the UCLA Transgenic Mouse Core and CSMC Mouse Genetics Core. JAX011011 established by Dr. Jaenisch, 34 containing four DOX-inducible mouse reprogramming genes (Oct4, Sox2, Klf4, and c-Myc: OSKM) from Col1a1 locus was purchased from the Jackson Laboratory (Bar Harbor, ME). Transgenic mouse carrying LCMV gp33-specific TCR (P14) was purchased from UCLA DLAM breeding colony service.
Generation of a mouse strain containing four DOX-inducible reprogramming genes
A novel mouse line [B6129P2/OlaHsd-Gt(ROSA)26 rtTA/Oct4-Klf4-Sox2-cMyc] abbreviated as B6/129OKSM] in which every somatic cell contains four DOX-inducible reprogramming genes was established as follows. AinV15 cells were differentiated into fibroblast-like cells by culturing in 10% FCS/DMEM for 2 weeks. Differentiation status was confirmed by fibroblast-like morphology and expression of fibroblast marker, ER-TR7. Cells were transduced with a polycystronic lentiviral vector encoding reprogramming genes (STEM-CCA vector obtained from Dr. Mostoslavsky 35 ) and replated on iMEF-feeder cells at day 7 after vector transduction. Reprogramming into iPSCs was induced by culturing those cells for 2 weeks in ESC medium under the presence of 1 μg/ml DOX. ESC-like colonies were selected and monitored for the expression of pluripotent stem cell markers (SSEA1 and Nanog) by standard fluorescence microscopy as well as flow cytometry. After 10 passages, three independent clones positive for those markers were selected and used for generation of chimeric mice to validate pluripotency. Resulting chimeric mice were then crossbred with B6 mice, and F1 hybrids with an agouti coat derived from the 129P2/OlaHsd strain were crossbred for >10 generations to establish an inbred mouse line carrying a single copy of the STEM-CCA insert in the same allele (i.e., homozygote) and enabling induction of ESC-like colony formation from peripheral blood cells with high efficiency in the presence of DOX. Integrated vector copy number was assessed as reported previously. 36 Insertional mutation of reverse tetracycline-controlled transactivator in ROSA26 loci was monitored by genotyping using oIMR8052, oIMR8545, and oIMR8546 primers as recommended by the Jackson Laboratory.
Somatic cell reprogramming of exhausted LCMV-specific CD8 T cells
Isolation of LCMV-specific CD8 T cells was performed as follows. Briefly, five B6/129OKSM mice were infected with LCMV CL13 to induce chronic infection. As a positive control, four additional B6/129OKSM mice were infected with LCMV Armstrong (ARM). 37 Splenocytes were isolated from the infected mice at days 45 or 22 after infection of LCMV CL13 or LCMV ARM, respectively. Triple-positive cells for anti-mouse CD3 and CD8 antibodies as well as LCMV gp33 tetramer, were flow sorted for reprogramming. The status of T cell exhaustion was confirmed by high levels of PD-1 expression and poor responsiveness to LCMV gp33–41 peptide pulsing (data not shown). Reprogramming of LCMV gp33-specific CD8 T cells was mediated by culturing cells on iMEF-feeder cells or Matrigel under the presence of 1 μg/ml DOX. To isolate homogeneous reprogrammed cell lines, single colonies with ESC-like morphologies, characterized by round colonies with a clear and bright border, 38 were passed onto six-well cell culture plates preplated with iMEF-feeder cells with no DOX. The single colonies were passaged every 2–4 days for >10 times until each clone had uniform ESC-like morphologies.
PCR analyses for immunoglobulin and TCR Dβ gene rearrangements
Gene rearrangements in immunoglobulin for B cells and TCR-β chain loci for T cells were analyzed by PCR using genomic DNA isolated from clones and a pair of specific primers. Genomic DNA was extracted with the DNeasy Kit (69504; QIAGEN, Hilden, Germany). One primer set for B cells (VHJ558 and J3) and three primer sets for T cells [TCRβ-D1 U-S & TCRβ-D1 U-A, Dβ2(E) & TCRβ-J2D-A, TCRβ-D1 U-S & TCRβ-J2D-A] were used to identify gene rearrangements of immunoglobulin and TCR-β chain, respectively. The PCR products were separated by gel electrophoresis.
Flow cytometry
Immunostained cells were acquired on LSRFortessa Flow Cytometer using FACSDiva software (BD Biosciences). Data were analyzed using FlowJo software (FlowJo, LLC, Ashland, OR). Cell sorting was performed by the CFAR Flow Cytometry Core Facility at UCLA using BD Aria III.
Results
Establishing B6/129OKSM mouse strain that exhibits high levels of reprogrammed colony formation from peripheral B and T cells
After the discovery of a somatic cell reprogramming technology by the transduction of four genes (Oct4, Klf4, Sox2, and c-Myc), various additional factors as well as induction methods have been developed and optimized. 39 –42,43 However, the generation of iPSCs is still inefficient, with just 1% of the original cell population forming iPSCs on average, and an even lower percentage forming T cells. 39,44,45
T cell exhaustion is a dysfunctional state of unresponsiveness to antigen stimulation, thus, reprogramming of exhausted T cells is anticipated to be challenging. Transgenic mouse strains with the four genes mentioned above have been developed and have shown successful reprogramming from rare blood cell populations. 34,45 One of the mouse lines, JAX011011, was tested for reprogramming of peripheral T cells, but the approach was unsuccessful. We attempted to establish a new mouse line that allowed highly efficient reprogramming of T cells isolated from peripheral blood (Fig. 1A). Briefly, fibroblast-like cells were generated by differentiating a mouse ESC line, AinV15 (ATCC:SCRC-1029) derived from 129P2/OlaHsdGt(ROSA)26SorrtTA mouse, then stably expressing reverse tetracycline transactivator (rtTA), and transducing it with a polycistronic lentiviral vector encoding the above four reprogramming genes with an order of Oct4, Klf4, Sox2, and c-Myc (OKSM) under the DOX-inducible promoter (STEM-CCA 35 ). Those cells were then plated on iMEFs and cultured for 2 weeks in ESC medium supplemented with 1 μg/ml DOX for induction of reprogramming gene expression. Three independent colonies exhibiting typical mouse ESC-like morphology were replated on iMEFs and selected based on stable expression of pluripotent stem cell markers (SSEA1, SSEA3, SSEA4, and Nanog) (data not shown) for chimeric mouse generation. A total of 23 chimera mice out of 28 live births were derived. The chimeric offspring manifested the distinctive mixed coat colors of the donor blastocysts (B6: black) and host genotypes (129P2/OlaHsd: mosaic coat colors of agouti, white, chinchilla, and black) (Fig. 1B). Generation of secondary iPSCs (2° iPSCs) 45,46 obtained by culturing cells originated from STEM-CCA vector-transduced AinV15 cells in the presence of DOX, was confirmed using tail-tip fibroblasts. ESC-like phenotypes of those 2° iPSCs were verified by the expression of SSEA1 and Nanog, and by differentiation in embryoid bodies (Fig. 1C, D). Three chimeric mice with high levels of coat color chimerism (>90%) were selected and bred with B6 mice for generation of F1 hybrids. F1 hybrids with an agouti coat were then crossbred for >10 generations to establish a stable mouse line carrying a single copy of the OKSM cassette, which was derived from STEM-CCA lentiviral vector, in the same locus homozygously, as well as inducing high levels of ESC-like colony formation from peripheral blood cells. This novel mouse strain was named as B6129P2/OlaHsd-Gt(ROSA)26rtTA/Oct4-Klf4-Sox2-cMyc (abbreviated as B6/129OKSM) following the rules for strain nomenclature of genetically engineered animals. 47
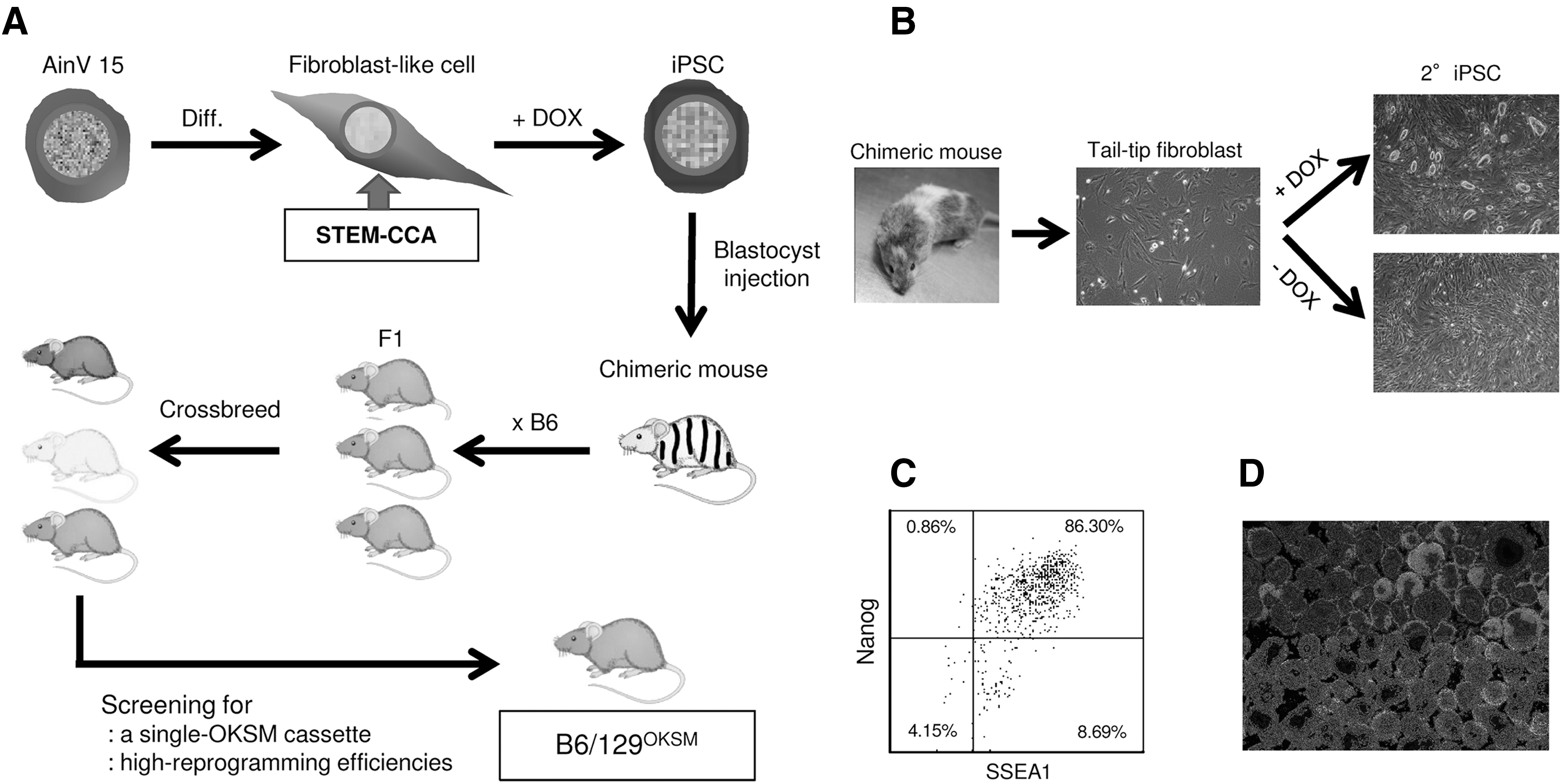
Establishment of B6/129OKSM mouse line.
We first compared reprogramming efficiencies of TTFs and peripheral blood cells obtained from our mice (B6/129OKSM) versus JAX011011 (Fig. 2). Reprogramming of TTFs and blood cells in the B cell expansion condition was successfully performed on iMEFs as reported previously. 38 This approach was unsuccessful for blood cells in the T cell expansion condition due to potent cytotoxic effects against iMEFs; most of the iMEFs were killed off within a week to 10 days and detached from the culture plate together with partially reprogrammed cells (data not shown). We, therefore, used plates coated with Matrigel for reprogramming of cells in the T cell expansion condition.

Highly efficient induction of 2° iPSC from peripheral blood cells of B6/129OKSM mice.
Successful ESC-like colony formation with TTFs was achieved with both mouse strains (Fig. 2A), but the reprogramming efficiencies of our new mouse strain was 10- to 100-fold higher compared with those of JAX011011 (Fig. 2B). Similarly, a great number of ESC-like colonies were confirmed with blood cells from our mice, but no colonies were seen with blood cells from JAX011011, in both culture conditions. We also tested the reprogramming efficiency in iMEF-conditioned medium, which would support efficient growth of reprogrammed cells, but there was no improvement (data not shown). To confirm whether blood cell-derived colonies originated from B or T cells, the rearrangement status of immunoglobulin heavy (IgH) locus and T cell receptor β (TCR-β) locus was assessed by PCR with genomic DNA using specific primer sets for each locus, respectively (Fig. 2C). Two out of five colonies in the B cell expansion condition contained rearranged IgH genes, whereas all five colonies in the T cell expansion condition contained rearranged TCR-β genes. Although we analyzed an additional 29 colonies in the B cell expansion condition, only 40% of total colonies (12 out of 29) contained rearranged IgH genes, indicating that other types of blood cells can be more efficiently reprogrammed compared to B cells in the B cell expansion condition. A strict comparison of reprogramming efficiencies of peripheral B and T cells was performed with magnetic bead-purified populations. The purity of each population was >99.5% (Fig. 2D). High levels of B and T cell reprogramming were seen with our mice, but no colony formation was observed with JAX011011. As expected, slightly higher levels of reprogramming were confirmed in the T cell population, especially with CD8 T cells, compared to the B cell population (Fig. 2E, CD4:CD8 = 0.9%:1.4% or 0.3%:0.9%).
Exhausted LCMV gp33-specific T cells successfully reprogram into iPSCs and redifferentiate into functional T cells maintaining original antigen specificity
The B6/129OKSM mouse strain described above allowed highly efficient reprogramming of blood cells, especially T cells, by simply culturing them in the presence of DOX. Using this mouse, we next determined whether exhausted T cells can reprogram into iPSCs while maintaining their original antigen specificity. B6/129OKSM mice were infected with either LCMV CL13, which induces chronic infection, or LCMV ARM, which causes acute infection 48 as a positive control. LCMV gp33-specific CD8 T cells were FACS sorted on day 22 for LCMV ARM and day 45 for LCMV CL13 after infection (Fig. 3A). Postsort analysis indicated that the sorting efficiencies were >99.0% in both cases. T cell exhaustion of the cells obtained from LCMV CL13-infected mice was verified by high levels of PD-1 expression and poor responsiveness to LCMV gp33-41 peptide pulsing (data not shown). Those cells were then reprogrammed on iMEF-coated and Matrigel-coated plates in the culture conditions described above.
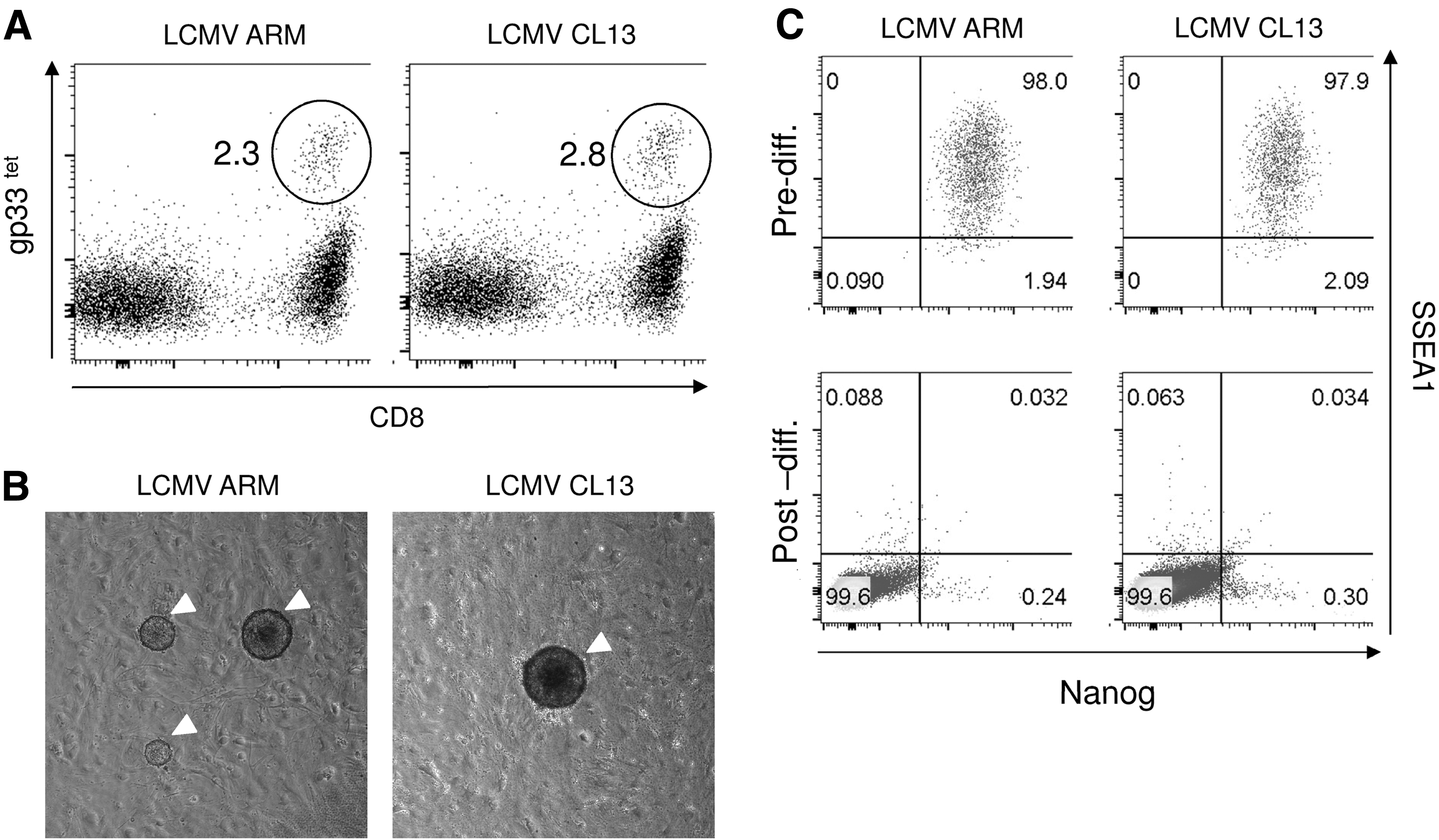
Reprogramming of exhausted LCMV-specific CD8 T cells.
ESC-like colonies appeared after around 3 weeks in both iMEF- and Matrigel-coated plates (Fig. 3B), but fewer colonies were seen in the latter (data not shown). Colonies double positive for SSEA1 and Nanog were selected and tested for their differentiation ability in embryoid bodies (Fig. 3C). Genomic DNA from each colony was extracted and processed for PCR-based TCR-β locus analysis (Fig. 4A, B). We successfully established 57 clones from T cells of LCMV ARM-infected mice and 33 clones from T cells of LCMV CL13-infected mice. Reprogramming efficiencies were much lower than those of pan CD8 T cells shown in Figure 2E; at 0.048% for LCMV ARM and 0.022% for LCMV CL13, respectively.
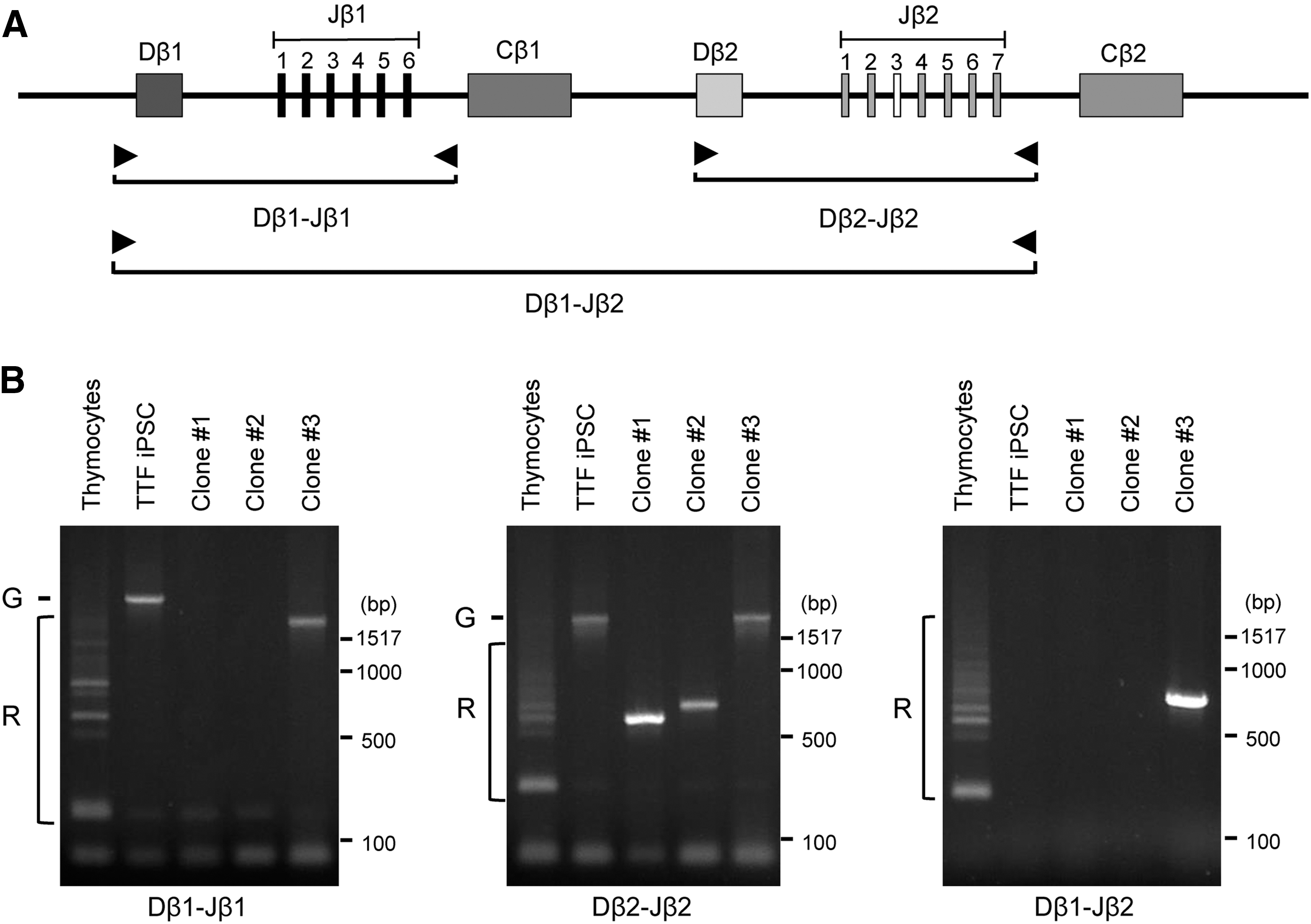
PCR-based analysis of TCR-β chain rearrangement in ESC-like clones. Total genomic DNAs were extracted from reprogrammed ESC-like clones established from exhausted LCMV gp33-specific T cells isolated from LCMV CL13-infected B6/129OKSM mice.
Three clones (Fig. 4B, Clone#1-3), derived from LCMV CL13-infected mice with different rearranged TCR-β patterns, were tested for their ability to differentiate into functional T cells while maintaining their original antigen specificity through chimeric mouse generation. To monitor progeny cells derived from the original cells used for injection into blastocysts, the clones were genetically labeled by an FG12 lentiviral vector encoding EGFP. 49 A total of 9 chimeric mice out of 54 live births were derived from those clones (2, 5, and 2 mice from clones #1, 2, and 3, respectively), indicating that they were derived from pluripotent precursors, that is, iPSCs. Overall, chimerisms, evaluated by visual assessment of coat color, were quite low compared with those from iPSCs originated from fibroblast-like cells transduced with STEM-CCA lentiviral vector (compare mouse pictures from Figs. 1B and 5A). Of those clones, peripheral CD8 T cells derived from clone #3 contained LCMV gp33-specific CD8 T cells [Fig. 5B, 3.0% in total CD3+/CD8+ T cell population (red box)]. As expected, EGFP+ cells in the CD3+/CD8+ population were also positive for LCMV gp33 tetramer (Fig. 5B), indicating that iPSCs derived from exhausted LCMV gp33-specific T cells were successfully redifferentiated into CD8 T cells that maintained their original antigen specificity. Importantly, those CD8 T cells positive for EGFP and LCMV gp33 tetramer expressed cell surface markers for naive T cells [CD62L+/CD44lo/CD25−, and CD127+(data not shown)] and no PD-1, which is one of the T cell exhaustion markers, similar to those in the EGFP population (Fig. 5C). These results indicate that exhausted LCMV gp33-specific CD8 T cells can reprogram into iPSCs and restore a naive T cell surface phenotype while maintaining their original antigen specificity. We could not confirm EGFP+ T cells in chimeric mice originated from clones #1 and #2, possibly due to low levels of chimerism from those clones (<0.1% EGFP+ cells in peripheral blood).
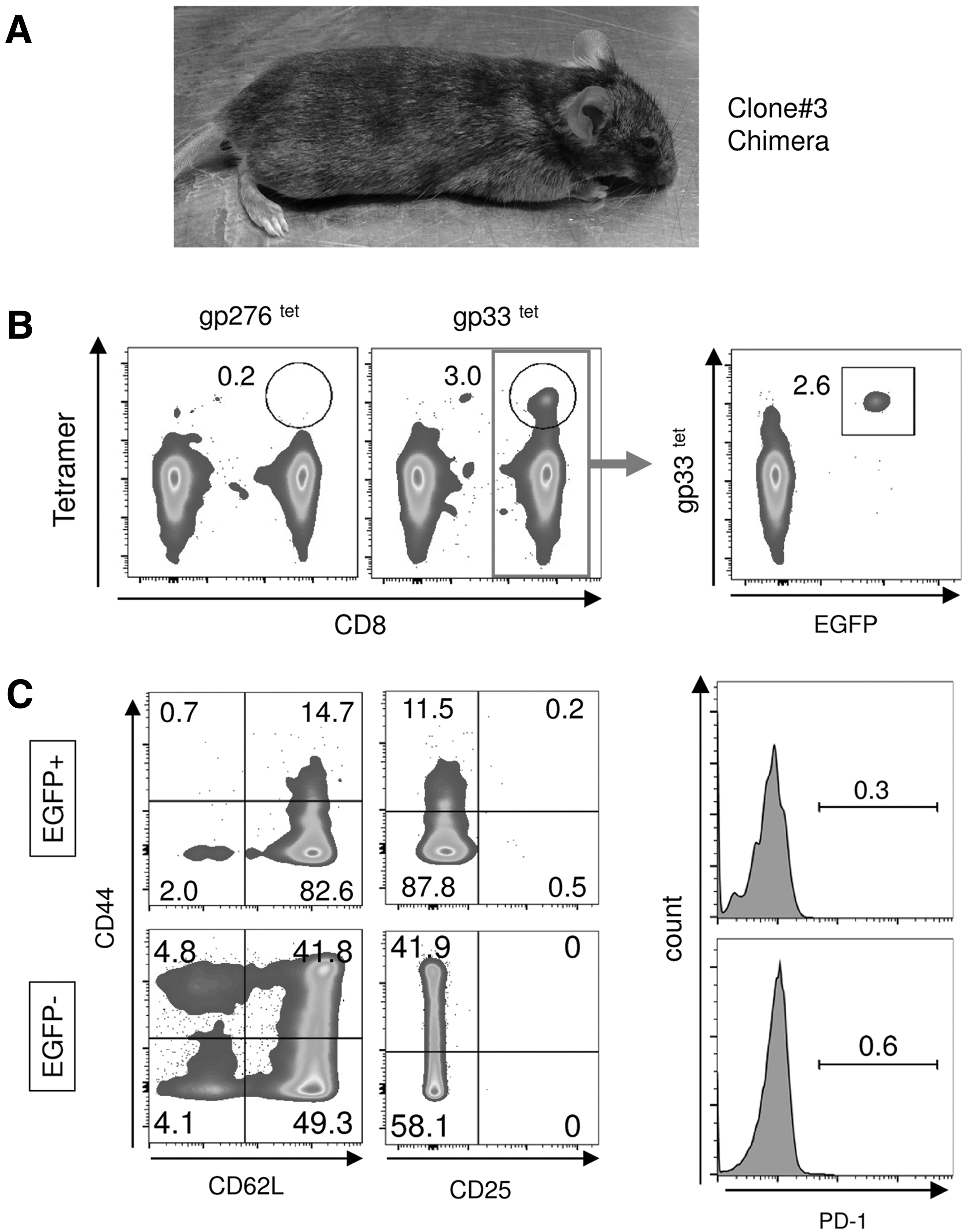
Redifferentiation of iPSCs derived from exhausted LCMV-specific T cells into T cells with a naive phenotype in vivo.
Antigen-specific T cell responses of those LCMV gp33-specific CD8 T cells were assessed by LCMV gp33-41 peptide pulsing in the presence of anti-mouse CD28 antibody, IL-7, and IL-15 (Fig. 6). Expansion of LCMV gp33-specific CD8 T cells was verified upon peptide pulsing [Fig. 6A, gp33 tetramer-positive cells in 106 CD3 T cells: 8,700 cells (prepulsing) vs. 110,000 cells (postpulsing)]. A majority of the LCMV gp33-specific CD8 T cells exhibited a surface phenotype of activated effector T cells (Fig. 6B, 40% PD-1+/CD25+). A similar observation was made in CD8 T cells from P14+ B6/129OKSM, which was generated by crossbreeding B6/129OKSM mice with transgenic mice carrying an LCMV gp33-specific TCR (P14) gene. 50 No or minimal levels of LCMV gp33-specific CD8 T cell response was detected from CD8 T cell populations that were negative for LCMV gp33 tetramer in the chimera or naive B6/129OKSM mice. In conclusion, these results indicate that exhausted LCMV gp33-specific CD8 T cells can reprogram into iPSCs and redifferentiate into functional naive T cells with their original antigen specificity.
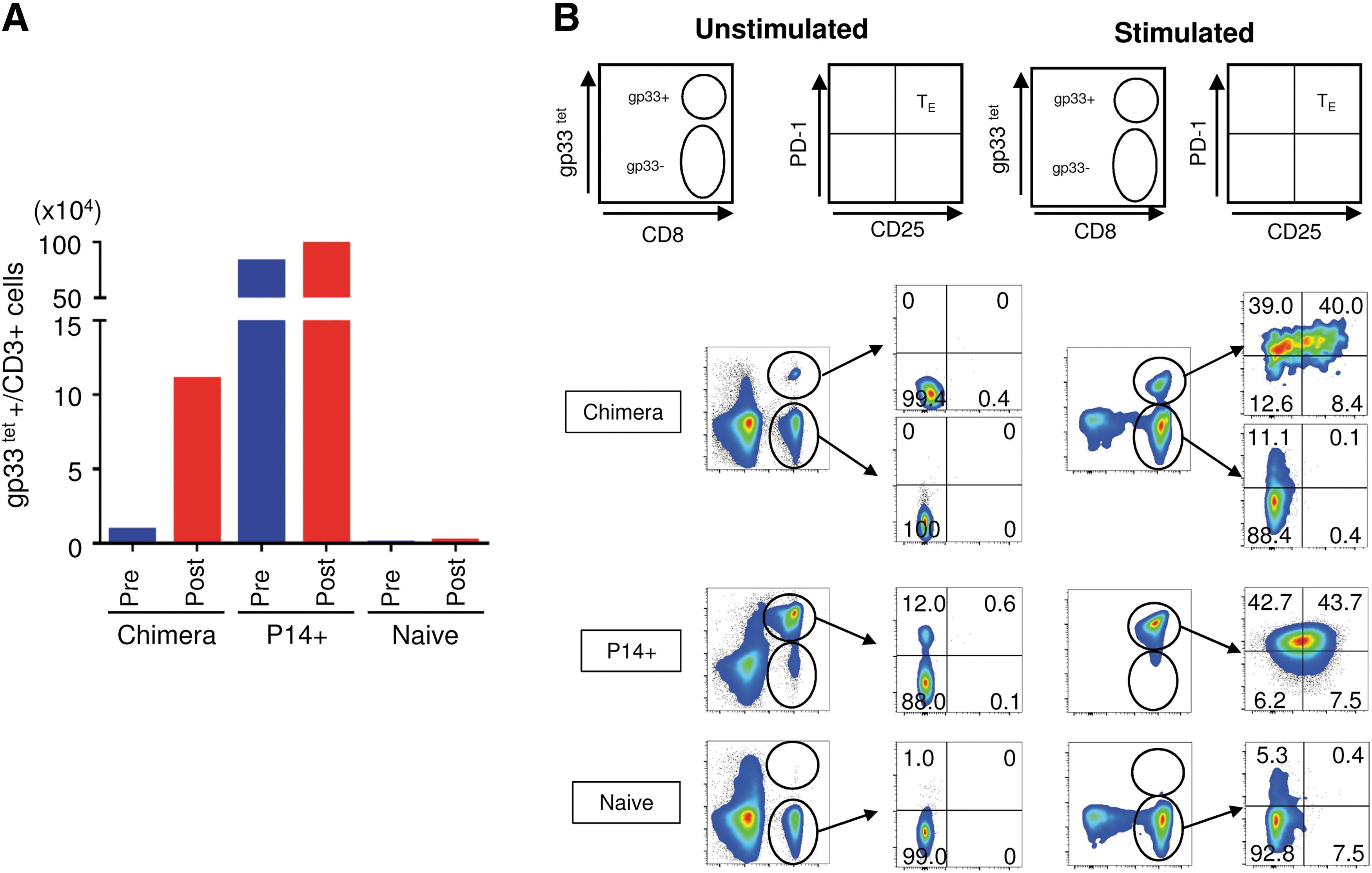
Antigen-specific activation of redifferentiated T cells from iPSCs originated from exhausted LCMV-specific T cells in vitro. Peripheral blood cells obtained from the chimeric mouse shown in Figure 5 (Chimera) were pulsed with 1 μg/ml of LCMV-derived gp33-41 peptide (KAVYNFATM) for 2 h in the presence of anti-mouse CD28 antibody (1 μg/ml). Cells were then cultured for 3 days in the presence of 5 ng/ml of IL-7 and IL-15.
Discussion
We assessed the feasibility of restoring functional effector T cell activity to exhausted antigen-specific CD8 T cells while maintaining their original antigen specificity, by a somatic cell reprogramming technology. Using a murine chronic infection model, our results indicate that (1) exhausted antigen-specific CD8 T cells can be reprogrammed into iPSCs, (2) those iPSCs can redifferentiate into CD8 T cells with a naive cell surface phenotype and parental antigen specificity, and (3) those T cells can functionally respond to antigen-specific stimulation. These results provide proof of concept for reversal of the exhausted T cell program by cleaning their previous dysfunctional state through a somatic cell reprogramming technology.
The mouse strain, B6/129OKSM, used in this research showed superior reprogramming efficiencies from peripheral B and T cells compared with JAX011011, which was developed by Dr. Rudolf Jaenisch with a similar concept. 34 Both strains contain single copies of four DOX-inducible mouse reprogramming genes, but the order of those genes is different. For our B6/129OKSM mouse strain, we used a STEM-CCA lentiviral vector to transduce a cassette encoding four reprogramming genes with an order of Oct4, Klf4, Sox2, and c-Myc (OKSM), whereas JAX011011 contains a different cassette with an order of Oct4, Sox2, Klf4, and c-Myc (OSKM). Comparative analysis of the JAX011011 strain 34 and another mouse strain carrying a single copy of the OKSM cassette (named the Collagen-OKSM reprogrammable mouse strain) developed by Dr. Konrad Hochedlinger, 51 both strains which contain a single copy of the reprogramming gene cassette in the same Col1a1 locus, indicated that the Collagen-OKSM reprogrammable strain had an increased mortality due to high tumor incidents. 52 In the B6/129OKSM strain, we did not observe similar abnormalities reported in the Collagen-OKSM reprogrammable mouse strain; eight incidents per 551 live births were confirmed in the B6/129OKSM strain within a 4-month period of observation. The B6/129OKSM strain was developed from three independent iPSC lines reprogrammed from fibroblast-like cells as shown in Figure 1A and used a STEM-CCA lentiviral vector to introduce an OKSM cassette, integration of which does not occur at specific genomic loci. 53,54 After over 10 generations of crossbreeding, we selected the strain that carried a single copy of the OKSM cassette and induced high iPSC formation from peripheral blood cells. We have confirmed that there were at least four independent integration sites in those iPSC lines used for chimeric mouse generation by PCR-based vector integration site analysis, 36 and that one of those sites located in chromosome six was maintained in the B6/129OKSM strain. Importantly, the high level of iPSC formation from peripheral blood cells was inherited even after backcrossing to B6 strain for over 10 generations (data not shown), suggesting that the integration site of the OKSM cassette is more important for efficient iPSC formation from blood lineage cells than the genetic background of the mouse strain. A detailed analysis of the relationship between reprogramming efficiency and the vector integration site is currently ongoing.
An adoptive transfer of functionally restored antigen-specific CD8 T cells to naive mice (i.e., cellular vaccine) or pre-infected mice (i.e., adoptive cell therapy) may be more practically relevant for clinical application. Such an attempt at adoptive transfer was unsuccessful due to insufficient LCMV-specific CD8 T cell development with our strategy using chimeric mouse generation (Fig. 5). We have no clear answer for this low donor chimerism observed with iPSCs generated from exhausted LCMV-specific CD8 T cells. As an alternative approach, we are currently attempting to differentiate those iPSCs into T cells in vitro using an OP9 coculture system. 55,56
The ultimate goal is to extend our studies into the clinical setting by reprogramming exhausted T cells specific to HIV proteins isolated from chronically HIV-infected patients. However, three technical advancements are needed for this research agenda: (1) reprogramming of exhausted antigen-specific T cells into iPSCs with exogenous reprogramming factors/conditions, (2) redifferentiate established iPSC lines into functional T cells in vitro, and (3) pre-expand those T cells to obtain a sufficient number of cells. All these technological advancements would make our concept a practical approach.
Authors' Contributions
M.K. designed the research, performed the experiments, analyzed data, and wrote the article. J.C., P.Y.K., E.K., Y.N., Y.L., J.W., H.J.E., M.Q., and G.E.R. performed the experiments. D.G.B. and I.S.Y.C interpreted data.
Footnotes
Acknowledgments
This work was supported by the California HIV/AIDS Research Grants Program (ID13-LA-563 to M.K.), UCLA AIDS Institute Mentorship Grant for Transitional Investigators (M.K.), NIH grant AI110200 (M.K.), NIH grant AI085043 (D.G.B), BSCRC Innovation Award (I.S.Y.C.), RS1-CIRM 00172-1 (I.S.Y.C), and Bill & Melinda Gates Foundation (I.S.Y.C). Cell sorting was performed in the CFAR Flow Cytometry Core Facility supported by NIH grants P30 CA016042 and 5P30 AI028697.
Tetramers specific to LCMV gp33-41 and LCMV gp276-286 were generated at the NIH Tetramer Facility.
Author Disclosure Statement
All authors declare no competing financial interests.