
Abstract
The genotypic properties of human immunodeficiency virus type 1 (HIV-1) subtype C in individuals presenting with cryptococcal meningitis (CM) are not well established. Employing single-genome amplification as well as bulk PCR, cloning and sequencing strategies, we evaluated the genetic properties of HIV-1 subtype C env in 16 antiretroviral therapy-naive study participants with CM. Eleven of the 16 participants had matched blood plasma and cerebrospinal fluid (CSF) evaluated, with the rest having either a plasma or CSF sample evaluated. Before antiretroviral therapy initiation, matched plasma and CSF-derived env sequences of all 11 participants displayed genetic intermixing between the two compartments. Overall, 7 of the 16 (∼43.8%) participants harbored CXCR4-using variants in plasma and/or CSF, according to coreceptor usage prediction algorithms. This study suggests that HIV-1 subtype C genetic intermixing between peripheral blood and the central nervous system is common in individuals presenting with CM, and that CXCR4 usage is present in one or both compartments in approximately 44% of individuals.
Introduction
T
HIV-1 is detectable in the central nervous system (CNS) in primary infection, 22 –24 where its presence may ultimately cause HIV-associated neurological disorders. 25,26 Studies of CNS compartmentalization of HIV-1 are limited because obtaining brain biopsies is not possible; however, the cerebrospinal fluid (CSF) has been used often as a surrogate of the CNS compartment. 27 –29 Early studies of HIV-1B env genes from matched plasma and CSF showed that variants unique to the CSF could arise, 30 and that this genetic compartmentalization of variants was most pronounced in individuals with HIV-associated dementia (HAD). 31,32 Later studies combining genotypic and/or phenotypic analyses confirmed clonal amplification of HIV-1B and HIV-1C variants in the CNS, indicating that the CNS can act as a compartment of unique HIV persistence relative to peripheral blood. 24,33,34 In a limited number of studies, a discordant entry phenotype of HIV-1B between the blood and CNS was reported in individuals with chronic stage HIV-1 disease. 27,35 –37 It is unclear how common genotypic discordance of HIV-1C between plasma and CSF is in individuals with advanced HIV infection and a coinfection.
Individuals harboring R5 HIV-1 only are eligible for combination antiretroviral therapy (cART) containing maraviroc, a CCR5 antagonist, while those harboring R5X4 or X4-tropic viruses are precluded from using this drug. 38 There are multiple genotypic tools or algorithms used to determine the coreceptor usage of HIV-1 to determine eligibility for use of maraviroc. 39 One approach is to use coreceptor usage prediction algorithms (CPAs) such as Geno2pheno, WebPSSM, and PhenoSeq, which utilize the whole V3 loop sequences of HIV-1 Envs to predict whether a virus will be R5 or CXCR4-using (using CCR5 and CXCR4, or CXCR4 alone). 40 –42 CPAs, therefore, are attractive diagnostic tools, as they are relatively inexpensive and less laborious to use than determinations by in vitro phenotypic assays.
HIV-related cryptococcal meningitis (CM) is an acquired immune deficiency syndrome (AIDS) defining illness, which is associated with high acute mortality rates, and is most prevalent in sub-Saharan Africa. 43 –45 Previous studies by our group revealed that compartmentalization of immune responses may arise between the blood and CNS compartment in individuals with CM, where proinflammatory natural killer cells, nonclassical monocytes, chemokines, and cytokines are enriched in CSF relative to blood. 46 We were interested in establishing whether or not HIV-1 genetic properties are compartmentalized in individuals with HIV-related CM. We hypothesized that localized inflammation in the CNS caused by Cryptococcus neoformans (or its antigens) provides an immunological milieu that recruits HIV-1C variants into the CNS, causing viral genetic mixing between peripheral blood and the CNS. We therefore explored whether CM was associated with HIV-1C genetic compartmentalization between blood plasma and the CSF or not, and whether in this setting there was evidence of CXCR4 usage by HIV-1 subtype C variants.
Materials and Methods
Study participants
The participants for this substudy were part of a larger prospective clinical study of cryptococcosis-associated immune reconstitution inflammatory syndrome (C-IRIS) conducted in Durban, South Africa, between August 2009 and September 2011, as described in detail elsewhere. 47 Written informed consent was provided by each participant or their next of kin. The ethics review boards of the University of KwaZulu-Natal (reference number BF053/09), Monash University (2009001224), and University of Western Australia (RA/4/1/2541) granted ethics approval for the study. For this study, 16 participants infected with HIV-1 and presenting with CM for the first time were randomly selected based on the criteria of detectable HIV-1 RNA in the plasma and CSF according to the COBAS TaqMan HIV-1 test (Hoffmann-La Roche, Basel, Switzerland) with a lower limit of 34 copies/mL, a CD4+ T cell count less than 200 cells/μL, and either a positive CSF cryptococcal antigen (CrAg) or Indian ink test result.
Plasma and CSF sample processing
The cell-free HIV-1 RNA levels in matched plasma and CSF samples were measured in the same polymerase chain reaction (PCR) assay run using the COBAS TaqMan HIV-1 test as described previously. 48 Routine cell count analyses, including the enumeration of white blood cell subsets in the CSF and CD4+ T cells in peripheral blood, were performed by standard clinical laboratory assays in an accredited laboratory. 47
RNA extraction and the synthesis of cDNA
The extraction of HIV-1 RNA from the plasma and CSF was performed using the QIAamp Viral RNA Mini kit (Qiagen, Dusseldorf, Germany) according to the manufacturer's instructions. SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA) was used for cDNA synthesis as previously described. 49 Briefly, 10 μL of extracted RNA was added to a mixture of 1 mM of deoxynucleoside triphosphates and 2 μM of primer OFM19 (5′-GCACTCAAGGCAAGCTTTATTGAGGCTTA-3′; HXB2 positions 9,604–9,632). The mixture was first heated to 65°C for 5 min and cooled to 4°C for 1 min. The following reagents were combined and then added to the initial mixture for a final volume of 20 μL per sample: 1 × reverse transcriptase (RT) buffer, 0.1 M dithiothreitol, 40 U of RNaseOUT (Invitrogen), and 200 U of SuperScript III Reverse Transcriptase enzyme. The complete reaction mixture was incubated at 50°C for 60 min, 55°C for 60 min to commence cDNA synthesis, incubated at 70°C for 15 min to inactivate the reverse transcriptase enzyme, and cooled to 4°C to end cDNA synthesis.
Single-genome amplification of plasma and CSF-derived variants
Unique viral variants were purified by single-genome amplification (SGA) and Sanger sequencing as described previously. 49 Synthesized cDNA was serially diluted, and a minimum of 10 PCRs for each dilution were performed. The dilution that presented less than or equal to 30% positive amplification reactions represented the endpoint dilution, where each positive amplification reaction contained a unique viral template. 50 Both rounds of PCR were performed using the Platinum Taq DNA Polymerase High Fidelity (Invitrogen) as described previously. 49,51 The first round of PCR included the forward primer VIF1 (5′-GGGTTTATTACAGGGACAGCAGAG-3′; HXB2 positions 4,900–4,923), reverse primer OFM19, and 1 μL of diluted cDNA. The second round of PCR included forward primer ENVA (5′-GCTTAGGCATCTCCTATGGCAGGAAGAA-3′; HXB2 positions 5,945–5,982), reverse primer ENVN (5′-CTGCCAATCAGGGAAGTAGCCTTGTGT-3′; HXB2 positions 9,145–9,171), and 2 μL of first-round PCR product. The cycling conditions for both rounds of PCR were as follows: 94°C for 4 min; 35 cycles of 94°C for 15 s, 55°C for 30 s, and 68°C for 4 min; and a final extension step of 68°C step for 20 min followed by a hold at 4°C. Positive PCRs were detected by agarose gel electrophoresis, and amplicons from endpoint dilution reactions were sequenced directly from the second-round PCR products.
Bulk PCR and cloning
Using a modified, nested bulk PCR and cloning protocol, 2.1 kb KpnI-to-BamHI env gene fragments were amplified from undiluted cDNA as described previously. 52 –54 Briefly, the forward VIF1 primer, reverse OFM19 primer, the Platinum Taq DNA Polymerase High Fidelity (Invitrogen), and 1 μL of undiluted cDNA were included in the first-round PCR mixture. In the second round of PCR, the forward Env-KpnI primer (5′-GTCTATTATGGGGTACCTGTGTGG-3′; HXB2 positions 6,336–6,359), reverse Env-BamHI primer (5′-GCTAAGGATCCGTTCACTAATCGT-3′; HXB2 position 8,463–8,485), the Phusion High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA), and 2 μL of PCR product from the first round of PCR were included. PCR products from second-round reactions were analyzed by agarose gel electrophoresis, and detected amplicons were gel purified. Purified amplicons were cloned into the pSVIII-Env plasmid for the purposes of sequencing. 52,54,55
Sequencing of HIV-1 env genes
SGA-derived amplicons from second-round PCR products and recombinant pSVIII-Env vectors were sequenced using the ABI Prism Big Dye Terminator Version 3.1 cycle sequencing kit (Applied Biosystems, Waltham, MA), incorporating multiple forward and reverse primers that covered the entire HIV-1 env gene. 56 Sequences were resolved using the ABI 3130 XL genetic analyzer and contigs composing env genes were assembled and manually edited with Sequencher version 5.4.1 (Genecodes, Ann Arbor, MI). Sequencing chromatograms with multiple overlapping peaks were discarded along with duplicate sequences.
Phylogenetic analyses
All participant-derived env genes were aligned with a panel of reference sequences from different subtypes available in the Los Alamos National Laboratory HIV database (
Compartmentalization analyses
For two participants, CM112 and CM089, there were a minimum of 10 SGA-derived sequences from the plasma and CSF each, allowing for evaluation of the degree of HIV-1 genetic compartmentalization between the two compartments as described previously. 33,58 Maximum-likelihood phylogenetic trees showing the intraparticipant phylogenetic relatedness of plasma and CSF-derived env genes were generated using MEGA 757 and PhyML software 59 with the following parameters: HK85 nucleotide substitution model, four substitution rate categories, a transition/transversion ratio of four, estimated gamma distribution parameter, and 1,000 bootstrap replicates. Compartmentalization of CSF sequences was determined using the Slatkin-Maddison (tree based) and Hudson nearest-neighbor tests (distance-based) with 10,000 permutations, in HYPHY as previously described. 33,58,60 A p-value equal to or lower than .05 signified CSF compartmentalization of sequences.
Prediction of coreceptor usage
The publicly available CPAs, Geno2pheno with a false positive rate of 5% (
Identification of N-linked glycosylation sites in the Env V3 loop sequences
Using the N-GlycoSite tool (
Net charge calculation
Positively charged amino acids (lysine, arginine, and histidine) were scored as +1, and negatively charged amino acids (aspartate and glutamate) were scored as −1. The net charge of the Env V3 was the sum of all positive and negative charges within the V3 sequence.
Statistical analyses
Two-tailed, unpaired nonparametric t-tests were performed using GraphPad Prism version 5 for Windows (GraphPad Software, San Diego, CA) unless otherwise stated.
Results
Clinical features of individuals harboring HIV-1 and presenting with CM
The detailed demographic and clinical characteristics of HIV-1-infected individuals with CM in the parent study have been described in detail elsewhere. 47 In this study, we randomly selected eight males and eight females infected with HIV-1 and presenting with CM, with matched plasma and CSF samples available for HIV-1 genotypic analyses. The samples were collected before initiation of cART. Demographic and clinical characteristics of these study participants are summarized in Table 1. The median age of the participants was 29.5 years (range: 23–45), with no significant difference between the median age of males and females (p = .46), and the median CD4+ T cell count was 27.5 (interquartile range: 10–72 cells/μL). The median plasma and CSF HIV-1 viral load of the participants were 5.1 (interquartile range: 4.72–5.2) log10 copies/mL and 4.73 (range: 4.3–4.7) log10 copies/mL, respectively, and were not significantly different (p = .13). There was no significant difference between male and female plasma HIV-1 viral load (p = .23), and no significant difference between male and female CSF HIV-1 viral load (p = .88). Two participants, CM032 and CM117, did not have detectable white blood cells in the CSF, whereas all other participants exhibited pleocytosis (>5 CSF white blood cells/μL). We observed a positive correlation between the CD4+ T cell count and white blood cell count in the CSF (spearman r = 0.7, p = .0026).
The identification number of the study participant.
The CD4+ T cell count was recorded as cells/μL.
The cell-free HIV-1 RNA viral load (VL) in plasma and CSF was recorded as log10 copies/mL.
The CSF white blood cell (WBC) count was recorded as cells/μL.
CSF, cerebrospinal fluid.
The phylogenetic relationships of HIV-1 variants
We amplified and sequenced a total of 251 unique env genes from the plasma and CSF compartments of 16 participants, using either SGA and sequencing, or bulk PCR, cloning and sequencing methods. The number of sequences generated per compartment of each participant using the two methods is summarized in Table 2. Phylogenetic analysis, performed as described in Materials and methods, revealed that all the sequences generated were subtype C. As expected, plasma and/or CSF sequences of each individual participant formed a unique cluster. All of the eleven participants with available matched plasma and CSF sequences had evidence of intercompartment mixing of env sequences (Fig. 1).

A neighbor-joining phylogenetic tree of participant-derived HIV-1 envelope gene sequences and a panel of reference sequences from different subtypes. All the sequences from individual study participants share the same color branches, and reference subtype sequences have gray branches. Closed circles and triangles at the tip of each branch indicate predicted R5 and CXCR4-using plasma-derived sequences, respectively. Open circles and triangles indicate predicted R5 and CXCR4-using cerebrospinal fluid-derived sequences, respectively. No interparticipant mixing of sequences was observed, suggesting an absence of interparticipant sequence contamination. Furthermore, all the participant-derived sequences clustered with reference subtype C sequences, while all other reference sequence subtypes clustered together, but separately from subtype C sequences. HIV-1, human immunodeficiency virus type 1. Figure 1 can be viewed in greater detail online at
The identification number of the study participant.
Sequences were generated by the single-genome amplification (SGA) and sequencing, or bulk PCR, cloning and sequencing method. Dashes indicate where we did not attempt to generate sequences for this sample using this method, and zeros indicate where we were unable to amplify cDNA before the sample was completely consumed or we could not amplify an env gene.
Genetic compartmentalization between anatomical compartments is more accurately determined using SGA-derived sequences than bulk PCR-derived sequences, as the former limits or eliminates Taq-induced recombination, nucleotide misincorporation, template resampling, and cloning bias. 49 We sought to establish whether there was genetic compartmentalization of HIV-1C sequences between the plasma and CSF or not of two participants (CM089 and CM112), where we generated at least 10 sequences from both of their compartments by SGA. For both participants, intercompartment mixing of env sequences was evident (Fig. 2). The Slatkin–Maddison test revealed seven and 10 intercompartment migrations in participants CM089 and CM112, respectively. In addition, neither CM089 nor CM112 demonstrated compartmentalization of env sequences (p-value .19 and .35, respectively). This result was confirmed with the Hudson nearest-neighbor test (p-value .34 and .58 for CM089 and CM112, respectively).
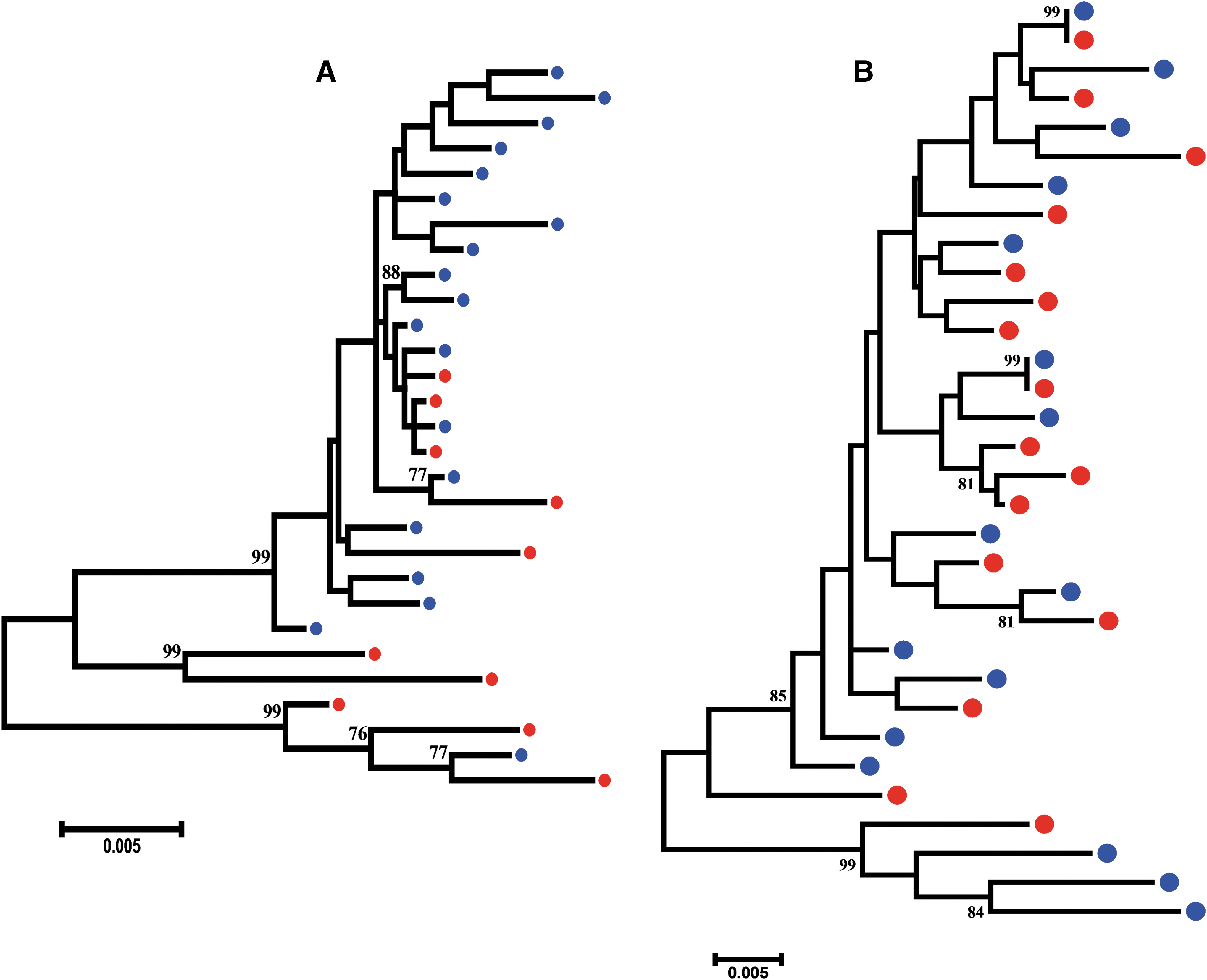
A maximum-likelihood phylogenetic tree of HIV-1 subtype C envelope variants in study participant CM089
Predicted coreceptor usage of HIV-1C in plasma and CSF
To get better insight into coreceptor usage by HIV-1 subtype C variants in end-stage disease, we used three publicly available CPAs (Geno2pheno, PSSM-C, and PhenoSeq) to generate a consensus prediction of the coreceptor usage of unique V3 sequences obtained in this study (Table 3).
The identification number of the study participant.
The name of a unique sequence; the first and second number in the round brackets indicate the number of other sequences with the same V3 in plasma and CSF, respectively, of the participant. The predicted coreceptor usage was determined using Geno2pheno (G2P, with false positive rate of 5%), PhenoSeq, and WebPSSM subtype C sinsi (PSSM-C). Consensus predicted tropism was determined based on the agreement of three, or two of three coreceptor usage prediction algorithms (CPAs). The amino acids occupying positions 11 and 25 (Pos. 11, 25) were identified using PSSM-C; a dash indicates a gap at that position. The number of potential N-linked glycosylation sites (PNGS) in the V3 sequence was counted using N-GlycoSite.
We also assessed whether predicted R5 variants predominate over CXCR4-using variants in plasma and CSF samples (Fig. 3). From the 14 plasma samples where we generated HIV-1 env genes, we identified that 7 of 14 (50%) participants harbored R5 variants only, whereas one participant (7.1%) harbored only CXCR4-using variants and six participants (42.9%) harbored a combination of R5 and CXCR4-using variants. Thus, based on the plasma samples only, R5 variants featured in 13 participants (92.9%) and CXCR4-using variants were detected in seven participants (50%). When we assessed the predicted coreceptor usage of variants in the 13 CSF samples where we generated sequences, we identified that nine of 13 (69.2%) CSF samples harbored R5 variants only, whereas two (15.4%) CSF samples each harbored R5 and CXCR4-using, and CXCR4-using variants only. Therefore, R5 variants featured in 11/13 (84.6%) CSF samples, while CXCR4-using variants were featured in 4/13 (30.8%) CSF samples. These results suggest that R5 variants predominate in plasma as well as CSF samples, and that CXCR4 usage is more common in plasma than in the CSF of HIV-1C-infected participants with CM. Overall, seven of the 16 (∼43.8%) participants included in this analysis harbored CXCR4-using variants in plasma and/or CSF.
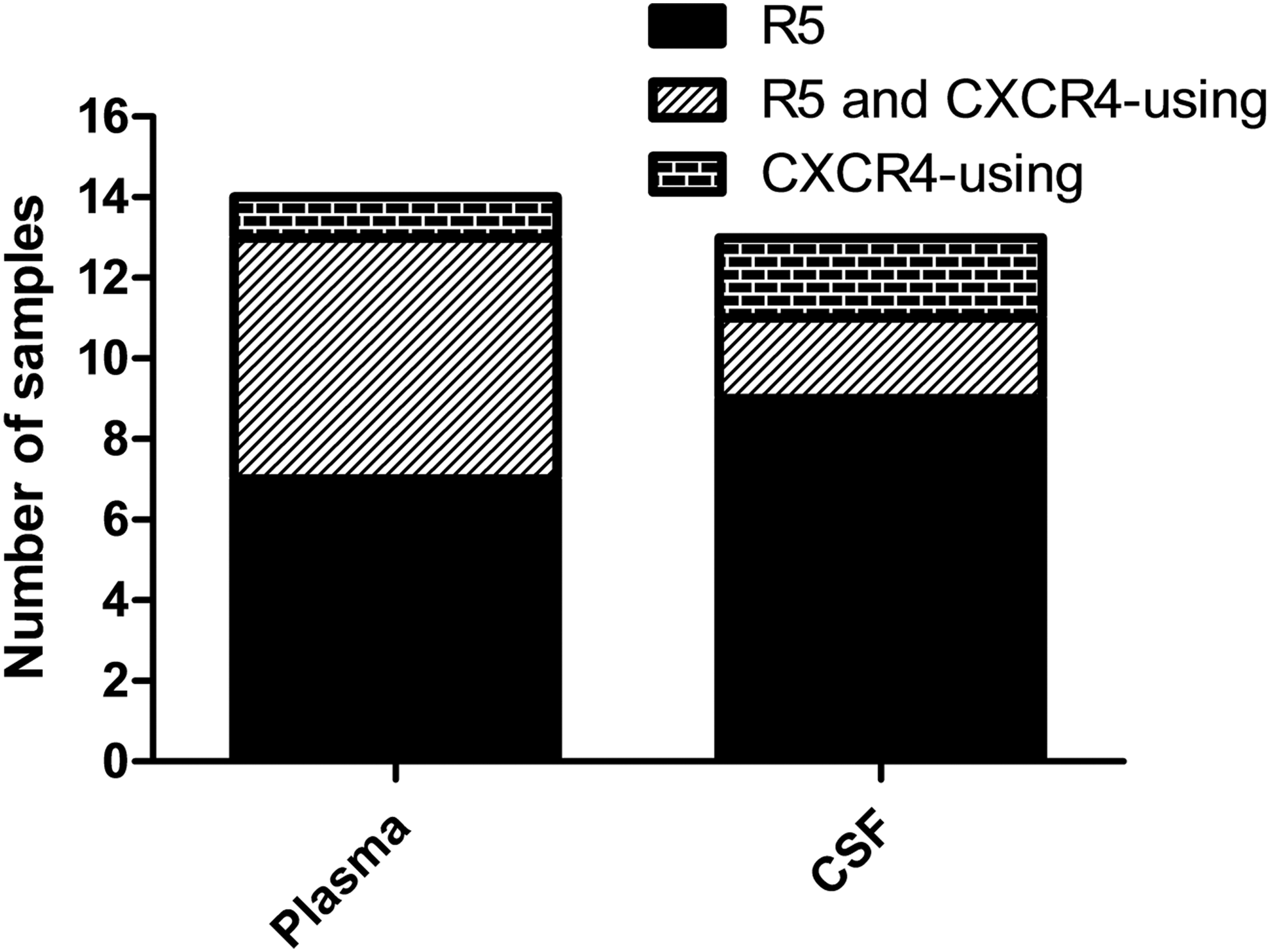
The predicted coreceptor usage in plasma and CSF samples. The number of plasma and CSF samples harboring R5, R5 and CXCR4-using, or CXCR4-using variants are shown as stacked columns. CSF, cerebrospinal fluid.
We next assessed whether matched plasma and CSF samples have HIV-1C variants with concordant coreceptor usage or not (Table 4). Seven of 11 (63.6%) participants with matched plasma and CSF sequences displayed concordant predicted coreceptor tropism of HIV-1C. A further 2 of 11 (18.2%) participants had discordance of predicted coreceptor tropism between plasma and CSF, where both R5 and CXCR4-using were detected in plasma, but only CXCR4-using or R5 variants were detected in CSF. In 2 of 11 (18.2%) participants (CM019 and CM032), the concordance of coreceptor usage between the plasma and CSF was indeterminate as we only had one CSF sequence compared to multiple plasma sequences. Our results show that concordant predicted coreceptor tropism between matched plasma and CSF compartments predominates over discordance in individuals with CM.
The identification number of the study participant.
The abbreviation “n.d” (not determined) indicates where we did not generate a sequence for the sample.
CM, cryptococcal meningitis.
Coreceptor usage determinants of HIV-1C Env
Understanding the determinants of CCR5 or CXCR4 usage by HIV-1C is of clinical relevance, given that a coreceptor antagonist, maraviroc, is FDA approved and available to individuals who harbor R5 variants only. 38,62 Previously, it was shown that HIV-1C CXCR4-using variants have specific Env third variable loop (V3) properties, which distinguish them from R5 variants. CXCR4-using HIV-1C variants have a V3 loop with greater amino acid variability, increased net charge, increased length, increased frequency of insertions, GPGQ crown motif substitutions, lower glycosylation site numbers, and charged residues at position 11 and/or 25 of the V3. 15,21,63 –66 Given that we identified a high prevalence of predicted CXCR4 usage by variants in this study, we sought to identify genetic properties of the major determinant of coreceptor usage, the V3 loop, which confers the ability to use CCR5 and/or CXCR4.
We determined the following additional properties of the V3 of all the sequences generated in our study: the overall net charge, number of potential N-linked glycosylation sites (PNGS), residues at position 11 and 25, and length (Table 3). Although 251 unique env genes were available in this study, only 58 unique V3 sequences were identified and aligned with a reference South African HIV-1C sequence (Fig. 4). The three CPAs all agreed on the predicted coreceptor usage for 48 of 58 (82.8%) unique V3 sequences, but in 10 of 58 (17.2%) instances, only two of the three CPAs agreed with each other.

An amino acid sequence alignment of unique HIV-1C envelope V3 sequences from plasma and cerebrospinal fluid-derived viruses of antiretroviral therapy-naive individuals living with CM. A reference South African HIV-1C envelope
As expected, the net charge of variants predicted to use CXCR4 (median 5; range 3–9) was significantly higher (p < .0001) compared with variants predicted to use CCR5 only (median 4; range 2–5) (Fig. 5). CXCR4-using variants of participants CM019, CM050, and CM052 did not have a positively charged residue at position 11 and/or 25 of the V3, whereas participants CM032, CM070, CM098, and CM108 harbored at least one variant with a histidine, arginine, or lysine at position 11 and/or 25. No variant predicted as R5 possessed a charged residue at position 11 and/or 25. This suggests that a charged residue at position 11 and/or 25 may be used to confer usage of CXCR4 for some CXCR4-using variants. In addition, all CXCR4-using variants (except three from participant CM032 and one from CM052) had a charge residue at position 16 and/or 18 of the V3, within the crown motif. R5 variants maintained a GPGQ crown motif generally.
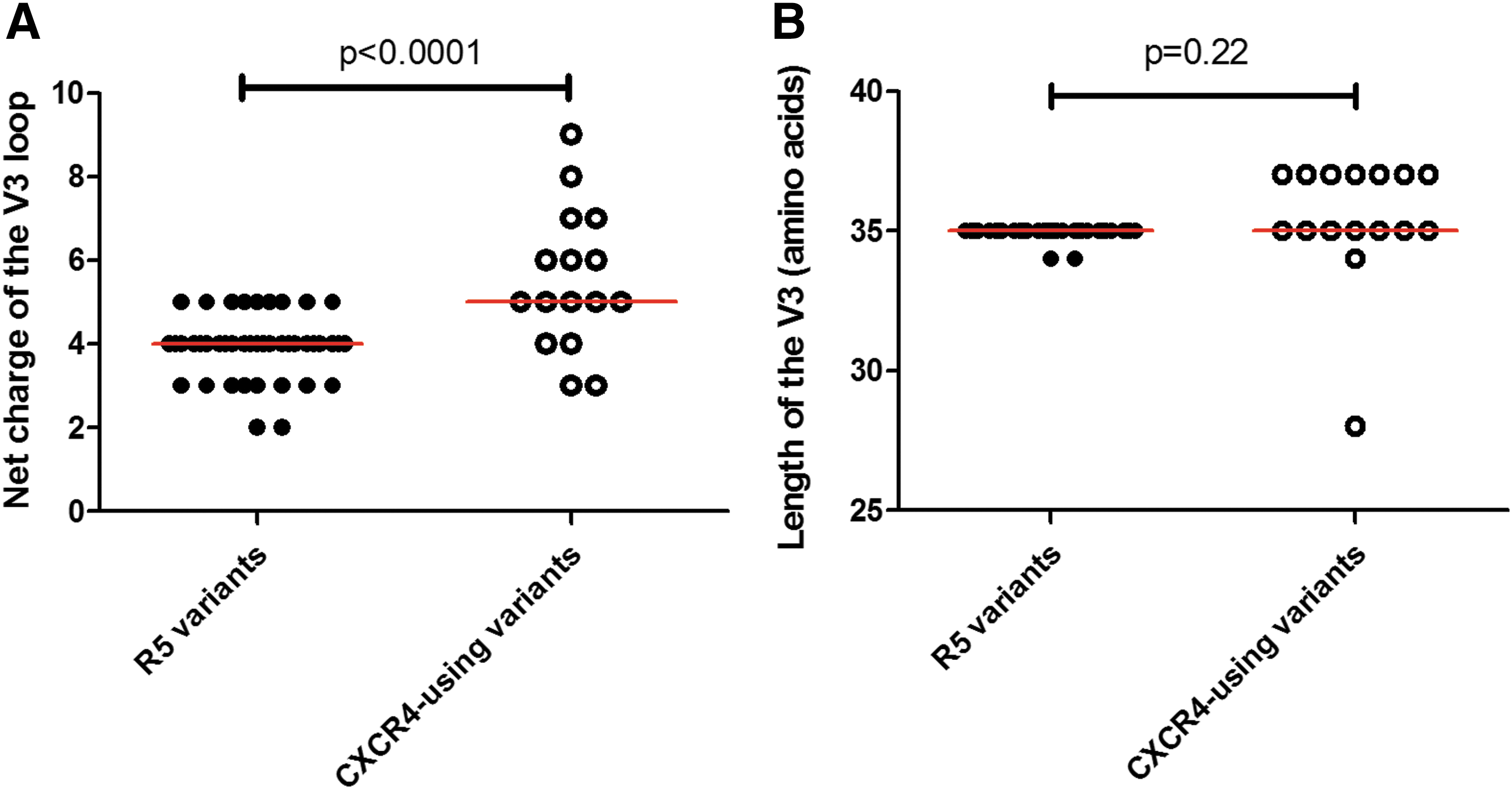
The net charge and length of HIV-1 subtype C envelope V3. The net charge
We then identified multiple sequences from participant CM019 and one from participant CM032, which did not have PNGS and were predicted to use CXCR4, while every other sequence had one PNGS only at position 6 of the V3. This suggested that the absence of a PNGS in the V3 may contribute to the usage of CXCR4, but is not compulsory for CXCR4 usage. The range of V3 length for CXCR4-using and CCR5 variants was 28–37 and 34–35 amino acids, respectively, both with a median of 35 (Fig. 5).
Rare mutation of the V3
The V3 loop of HIV-1 Env is a region important for function and maintains signature sequences according to subtype. The isoleucine at position 14 of the V3 (position 309 according to HXB2 numbering or I309) is important for the structure of the V3 loop, and is conserved in approximately 99% of HIV-1C sequences.
67
I309 also modulates the exposure of the CD4 binding site.
68
Notably, five unique V3 loop sequences of participant CM108 had a rare replacement of I309 with a phenylalanine residue, while every other sequence analyzed had an isoleucine at that position, including the reference South African HIV-1C sequence
Discussion
Although others have studied HIV-1C env genes in individuals with advanced HIV disease and AIDS-defining illnesses, 15,17,64,69 genetic information of HIV-1C in matched blood and the CNS compartments of individuals with CM is sparse. In this study, we characterized the phylogenetic relatedness, and predicted coreceptor usage of plasma and CSF-derived HIV-1 variants in individuals with CM.
The sampling of HIV-1 from the CSF has been shown to be an adequate surrogate of sampling virus from the brain. 29,70 Equilibration, partial or complete, and compartmentalization of viral sequences between plasma and CSF have been described previously. 24,30,33,34 The occurrence of extreme genotypic compartmentalization of HIV-1 in the CNS, in particular, is suggestive of independent replication and evolution of viruses relative to the blood, and this is strongly associated with the development of neurocognitive impairment. 31 –33 The implications of equilibration or compartmentalization of HIV-1 sequences between blood and the CNS are unclear for individuals with co-infections. In this study, we observed frequent intermixing of HIV-1C plasma and CSF-derived sequences in all 11 participants, such that plasma and CSF sequences did not form separate clusters, but were phylogenetically interspersed among each other. In addition, using the Slatkin–Maddison and Hudson nearest-neighbor compartmentalization algorithms on sequences from two participants CM089 and CM112, we observed an absence of compartmentalization of CSF-derived sequences in both participants. The majority of the participants in this study exhibited pleocytosis, a marker of CNS inflammation, which is associated with HIV-1 trafficking between peripheral blood and the CNS 71 ; therefore it is probable that Cryptococcus neoformans and/or HIV-1C co-infection led to a breach of the blood–brain barrier, which allowed movement of viruses between the peripheral blood and CNS compartments. Participants CM032 and CM117 did not display pleocytosis, however, although they had intercompartment mixing of sequences, suggesting that pleocytosis is not a prerequisite for genetic intermixing between blood and the CNS. Because we did not evaluate longitudinal samples, we also could not rule out a previous pleocytosis event that may have led to an absence of viral genetic segregation in these participants. Nevertheless, these results supported our hypothesis, where we expected that intercompartment mixing would arise in some individuals with CM. It is possible that immunological compartmentalization was a recent event associated with the development of active cryptococcosis, and therefore did not allow enough time for detectable independent viral evolution.
R5 variants establish infection in the CNS compartment, 23,24 but R5, R5X4, and X4 have been isolated from the brain later in disease. 72 We observed that concordance of HIV-1 coreceptor usage predominated in matched plasma and CSF samples. Our results were in agreement with other studies where phenotypic assays 35,36 and CPAs 37 were employed, showing that concordance of HIV-1 coreceptor usage predominated, but discordance of HIV-1 between the plasma and CSF occurred in 10.9%–35.7% of matched samples. In addition, we showed that plasma may harbor CXCR4-using variants, while the CSF harbors R5 variants only as described previously. 36,37 These results suggest that discordance of coreceptor usage between compartments was reflective of the functional compartmentalization of HIV-1, as others have shown that env sequences in the CNS may be compartmentalized. 73
Previously, it was shown that CXCR4-using HIV-1C has greater amino acid variability, increased net charge, increased length, increased frequency of insertions, GPGQ crown motif substitutions, lower glycosylation site numbers, and charged residues at position 11 and/or 25 of the V3 relative to R5 variants. 21,63 –66 In this study, we investigated whether the HIV-1C R5 and CXCR4-using variants predicted by CPAs possessed the same determinants of coreceptor usage as variants from previous studies. We identified that many previously described V3 properties specific to HIV-1C R5 and CXCR4-using variants are consistent with variants in our study. In summary, the predicted CXCR4-using variants in our study had, compared to R5 variants, V3 sequences with higher net charge, two amino acids insertions upstream of the crown motif, an absence of PNGS, a charged residue at position 11/and 25, and alterations of the GPGQ crown motif.
Changes in position 14 of the V3 (position 309 according to HXB2 numbering) affect the structure of the loop 67 and the exposure of the CD4 binding site. 68 We identified that sequences from participant CM108 possessed a change of the conserved isoleucine at V3 position 14 (I309) to phenylalanine (F309). Furthermore, the F309 residue was present in variants predicted as R5 and CXCR4-using, suggesting that this substitution was not unique to any particular coreceptor usage phenotype. Given that I309 was shown to influence the structure of the V3 and modulate the exposure of the CD4 binding site, it suggests that variants in participant CM108 may have a unique entry and immune evasion profile compared to variants with the I309 residue harbored by other individuals with CM.
Reliable estimates of the prevalence of CXCR4 by HIV-1 are required to understand the evolution of the HIV-1C epidemic and inform effective therapeutic interventions. HIV-1B has been shown to expand its usage of coreceptors in chronic and the end stages of disease. 10 –13 On the other hand, HIV-1C has been observed to switch major coreceptor usage less frequently. 14,15,19 –21 In some cases, a complete absence of CXCR4 usage by HIV-1C was observed in individuals with advance disease, suggesting that the ability to switch major coreceptors for this subtype is rare or not essential. 17,18 We showed that R5 variants predominate in both plasma as well as CSF samples, but that at least one CXCR4-using variant was present in plasma and/or CSF in ∼43.8% of study participants. Although we expected to detect CXCR4 usage by HIV-1C in some individuals in this study, considering that all had an AIDS-defining condition, we observed a much higher frequency of CXCR4 usage than expected, based on previous studies. It may be that the CPAs overestimated the predicted CXCR4 usage as described previously. 74 –77 Coreceptor usage tests in vitro may be employed in future to corroborate our findings. It remains unclear, however, whether CXCR4-using variants are enriched in individuals, depending on the co-infection they have, or that HIV-1C has evolved to use CXCR4 more over time. We also showed that 4/13 (30.8%) CSF samples harbored predicted CXCR4-using HIV-1C variants, indicating that different phenotypes besides R5 variants may circulate in the CNS in chronic to end-stage disease. The clinical importance of our findings may require further longitudinal evaluation, considering that genetic intermixing between peripheral blood and the CNS may afford HIV-1 the ability to target cells of the CNS using CXCR4.
Our study was limited to a small cohort of 16 participants with highly similar clinical characteristics. We did not have a control group of participants infected with HIV-1, but not presenting with CM. We generated a low number of sequence for one or both compartments of some participants; therefore, we may have missed minority lineages that are specific to the peripheral blood or CNS compartment. Another limitation of the study, which may have impacted the results of our study, was the use of bulk PCR amplification and cloning together with SGA to generate sequences. SGA has been shown to provide a better measure of viral variants, especially minority ones, in a quasispecies compared to bulk PCR and cloning, as described previously. 49 On the other hand, a different study showed that bulk PCR and cloning may capture viral diversity as SGA does. 78
Future longitudinal studies assessing differences of HIV-1 in blood and the CNS of individuals with and without CM will improve our understanding of the pathogenesis of HIV-1. Our results suggest that trafficking of HIV-1 variants between blood and the CNS is common, although the mode and the effect on HIV-1 neurotropism is not completely clear. We also established using CPAs that maraviroc use would not be suitable for approximately 44% of individuals with CM, suggesting that an alternative mode of therapy must be optimized for them if standard regimens fail.
Footnotes
Acknowledgments
This research was funded by the Howard Hughes Medical Institute, the South African Department of Science and Technology/National Research Foundation Research Chairs Initiative and the Victor Daitz Foundation. Partial funding was provided by the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative [grant # DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS) Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency), with funding from the Wellcome Trust [grant # 107752/Z/15/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government. We extend our gratitude to Professor Paul Gorry, head of the HIV Molecular Pathogenesis Laboratory (Burnet Institute), for providing us with the pSVIII-Env plasmid. Finally, we thank and acknowledge the study participants.
Sequence Data
Nucleotide sequences are available in GenBank with the following accession numbers: MF284812-MF284921 and MF284922-MF285062.
Author Disclosure Statement
The authors have no competing interests.