
Abstract
Efficiency of artificial restriction enzymes toward curing HIV has only been separately examined, using differing delivery vehicles. We compared the in vitro transduction and target-mutagenesis efficiency of consortium plasmid and adenoviral vector delivered HIV-1 pol gene targeting zinc finger nuclease (ZFN) with CRISPR/Cas, Custom-ZFN, CRISPR-Cas-9, and plasmids and vectors (murCTSD_pZFN, pGS-U-gRNA, pCMV-Cas-D01A, Ad5-RGD); cell lines (TZM-bl and ACH-2/J-Lat cells); and the latency reversing agents prostratin, suberoylanilide hydroxamic acid, and phorbol myristate acetate. Cell lines were grown in either Dulbecco's modified Eagle's medium or Roswell Park Memorial Institute with the antibiotics kanamycin, zeocin, and efavirenz. Efficiency was assayed by GFP/luciferase activity and/or validated by yeast MEL1 reporter assay, CEL1 restriction fragment assay, and quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR). Ad5-RGD vectors had better transduction efficiency than murCTSD and pGS-U-gRNA/pCMV-Cas-D01A plasmids. CRISPR/Cas9 exhibited better target-mutagenesis efficiency relative to ZFN (delivered by either plasmid or Ad5 vector) based on gel electrophoresis of pol gene amplicons within ACH-2 and J-Lat cells. Ad-5-RGD vectors enhanced target mutagenesis of ZFN, relative to murCTSD_pZFN plasmids, to levels of CRISPR/Cas9 plasmids. Similar reduction of luciferase activity among TZM-bl treated with Ad5-ZFN vectors relative to CRISPR/Cas-9 and murCTSD_pZFN plasmids was observed on challenge with HIV-1. qRT-PCR of HIV-1 pol gene transcripts affirmed that Ad5 (RGD) vectors enhanced target mutagenesis of ZFN. Whereas CRISPR/Cas-9 may possess inherent superior target-mutagenesis efficiency; the efficiency of ZFN (off-target toxicity withstanding) can be enhanced by altering delivery vehicle from plasmid to Ad5 (RGD) vectors.
Introduction
I
In lieu, several strategies aimed at depleting the latent HIV-1 reservoir have been devised. 17 –34 This by either (1) reactivating latent virus and thereby accelerating its clearance or (2) targeting latently infected resting memory CD4+ T cells for destruction. 18 –34 A limitation for the first strategy is that it does not eliminate the provirus, while the second faces the difficulties, uncertainties, and potential dangers inherent in targeting the latently infected cells themselves.
An attractive approach would be to selectively disrupt integrated provirus already established within those latently infected cells. Eriksson et al. 35 observed that some of the resting memory CD4+ T cells already contain defective viral genomes, which do not express viral genes in response to latency reversing agents. Sanchez et al. 36 independently demonstrated an accumulation of defective viral genomes in peripheral blood monocyte cells of HIV-1-infected individuals. Kieffer et al. 37 specifically revealed G-to-A hypermutation in protease and reverse transcriptase (RT) regions of HIV-1 residing in resting CD4+ T cells in vivo. The human genome is generally comprised in large parts of remnants of ancient retroviral infections, which do not cause disease because they have effectively been rendered functionally defective. These data—in addition to an effective cocktail of both RT, integrase and protease inhibitors used in current HAART combinations, provide circumstantial evidence to support the postulate that induction of target mutagenesis among integrated HIV-1 genomes (or at least some of their genes) within resting memory CD4+ T cells can render latent provirus nonreactivatable.
Our research group 38,39 —more than 17 years ago, conceived that the natural antiviral innate defense system, through which bacteria inactivate bacteriophages through target mutagenesis, offered a model for an antiviral gene therapy against HIV. The potential for genome toxicity emanating from the short palindromes targeted by bacteria restriction enzymes (REases) 40 led us to harness artificial enzymes, starting initially with zinc finger nucleases (ZFNs) and more recently CRISPR/Cas. The unique hypothesis of our program is that, “ZFN or CRISPR/Cas-mediated target mutagenesis of vital segments of HIV-1 DNA can inactivate latent provirus and abort or restrict its replication.” A specific postulate that our team is now investigating is that, “target-mutagenesis and functional abrogation of the HIV-1 pol gene alone (encoding three critical enzymes for HIV replication: RT, integrase, and protease) effectively inactivate latent HIV-1 provirus.” 40 This would leave remnants of HIV-1 provirus that do not cause disease because they have been rendered functionally defective. 41 –52 The laboratory of Dr. H. Zhu at Fudan University in Shanghai, China, 53 has derived proof of concept for the alternate option of excising proviral HIV-1 from latently infected cells. Until now, the efficiency of artificial restriction enzymes (AREs) toward curing HIV has, however, only been separately examined, using differing delivery vehicles. In this study, we compared the in vitro transduction and target-mutagenesis efficiency of consortium plasmid and adenoviral vector delivered HIV-1 pol gene targeting ZFN with CRISPR/Cas-9.
Materials and Methods
Design: in vitro studies
Materials and supplies
Custom HIV-1 pol gene targeting ZFN and CRISPR-Cas-9 (from Sigma-Aldrich, GER, and GenScript, respectively); plasmids and vectors (murCTSD_pZFN, pGS-U-gRNA, pCMV-Cas-D01A, and Ad5-RGD); cell lines (TZM-bl and ACH-2/J-Lat cells from the NIH AIDS Reagent Program); and latency reversing agents prostratin (PST), suberoylanilide hydroxamic acid (SAHA), and phorbol myristate acetate (PMA) (obtained as gifts from GlaxoSmithKline, United Kingdom) were used. Details of materials, supplies, and all other utilities are described in the interventions.
Interventions
Synthesis of the three study target-mutagenesis AREs (i.e., ZFN and CRISPR/Cas-9), plasmids, or vectors
HIV-1 pol gene targeting ZFN and CRISPR/Ca9 along with their corresponding plasmids and vectors were engineered by subconstruct to Sigma-Aldrich, GER, and GenScript, respectively. The murCTSD_pZFN plasmid was hence lent to Vector Biolabs to generate ZFN carrying rugged adenoviral vectors, subtype 5 (Ad5-RGD). 40,54 –60
Cell lines, in vitro cell cultures, transductions, and luminescent or fluorescent marker assays
In anticipation of this work as early as August 2012, we obtained a material transfer agreement between our team at Makerere University and the U.S. NIH AIDS Research and Reference Reagent Program (ARRRP). The three cell lines from the NIH-ARRRP were grown in either Dulbecco's modified Eagle's medium (DMEM) or Roswell Park Memorial Institute (RPMI) impregnated with the antibiotics kanamycin and zeocin (to select for transduced cells, since the plasmids and vectors contained a resistance locus to these antibiotics) and efavirenz (to restrict excessive viral outgrowth). TZM-bl cell lines offered us a model for active HIV-1 infection, while ACH-2 and J-Lats offered us a model for latent HIV infection and reactivation. 61 –68 Transfection was done at 0.1, 0.2, 0.4, and 0.8 μl of the original volume after treatment with 0.2 μl of transfection reagent (Invitrogen). TZM-bl cells were further challenged with 1, 2, 4, and 6 μl of supernatants of HIV-1 clones derived from reactivation of ACH-2 cells. ACH-2 and J-Lat cells—that offered us a model for latent HIV infection and reversal, on the contrary, were activated with varying concentrations, that is, between 0.0002 and 1 μM/ml of any of the three latency reversing agents alone or in combinations of two or even three: PST, PMA, and SAHA (provided as a gift from GSK: manufactured by Sigma-Aldrich and Cayman Chemicals). Once more, to prevent cytotoxicity in cultures, efavirenz (also a gift from GSK: Cayman Chemicals) was included within the DMEM or RPMI culture medium at this stage. The luciferase assay in J-Lat cells was validated as described elsewhere. 69 –73
Target-mutagenesis validation by polymerase chain reaction amplicon digestion using a CEL1 assay in ACH-2 and J-Lat
Following treatment with latency reversing agents, both treated and untreated negative control isolates of ACH-2 and J-Lat cells were obtained at one, centrifuged and sediments processed for Promega
ZFN, zinc finger nuclease.
Quantitative RT-PCR to examine extent of mutagenic viral reactivation
Quantitative RT-PCR (qRT-PCR) was achieved using primers #4 and 5 alongside probe 6 (Table 1): (1) Extracting total genomic mRNA and converting it into cDNA. Cell cultures were centrifuged and the cell pellet harvested from which total RNA was extracted using the RNeasy
Measurable variables
(1) Plasmids and viral vectors were validated by gel electrophoresis, sequencing, and/or yeast MEL1 assay. (2) Live cell cultures were visualized and affirmed by microscopy and apoptotic assays. Transduction efficiency was qualitatively assayed by fluorescent microscopy of GFP marker inbuilt within the plasmid and vector. (3) Target-mutagenesis efficiency was examined by CEL1 SMAs on HIV-1 pol gene amplicons of ACH-2 and J-Lats. Abrogation of HIV-1 infectiousness and reactivation was, respectively, examined by fluorometry among TZM-bl cells and qRT-PCR of viral copies (vc/ml) among ACH-2 and J-Lat cells.
Data analyses
All experiments were repeated more than three times, and the best results are presented. Appropriate controls for each were used as described. Data were manually curated and analyzed on computer. For qRT-PCR, viral loads were computed as an inverse factor of CTs using a standard curve generated in-house.
Results
Synthesis of the three study target-mutagenesis AREs (i.e., ZFN and CRISPR/Cas-9) plasmids, or vectors
ZFN and consortium mammalian plasmids
A pool of six paired CompoZr™ knockout ZFNs (PZFN1/PZFN2; PZFN3/PZFN4; and PZFN5/PZFN6) were computationally derived against the HIV-1/SHIV pol gene target sequences (Fig. 1a). The issuing individual ZFN was cloned and tested for mRNA/DNA quality (Table 2), while the paired sets were tested for in vitro target-mutagenesis efficiency using a yeast MEL1 reporter assay (Fig. 1b). Although all three pairs demonstrated >50% relative MEL1 to positive control activity, the first pair or set 1 (PZFN1/PZFN2) offered the most promising efficient target mutagenesis in vitro (Table 3). To enhance target cell transduction efficiencies, each of the validated ZFN of a paired set was individually subcloned into a separate consortium mammalian plasmid (see Fig. 1c for plasmid-vector map construct). This work was done by way of subcontract to Sigma-Aldrich (GER). The delivered plasmids at Makerere University were subcloned into Escherichia coli strain DH5Alpha for further propagation. The DNA quality of the expanded plasmids by both NanoDrop and gel electrophoresis is shown in Table 4 and Figure 1d, respectively. 40,54 –56
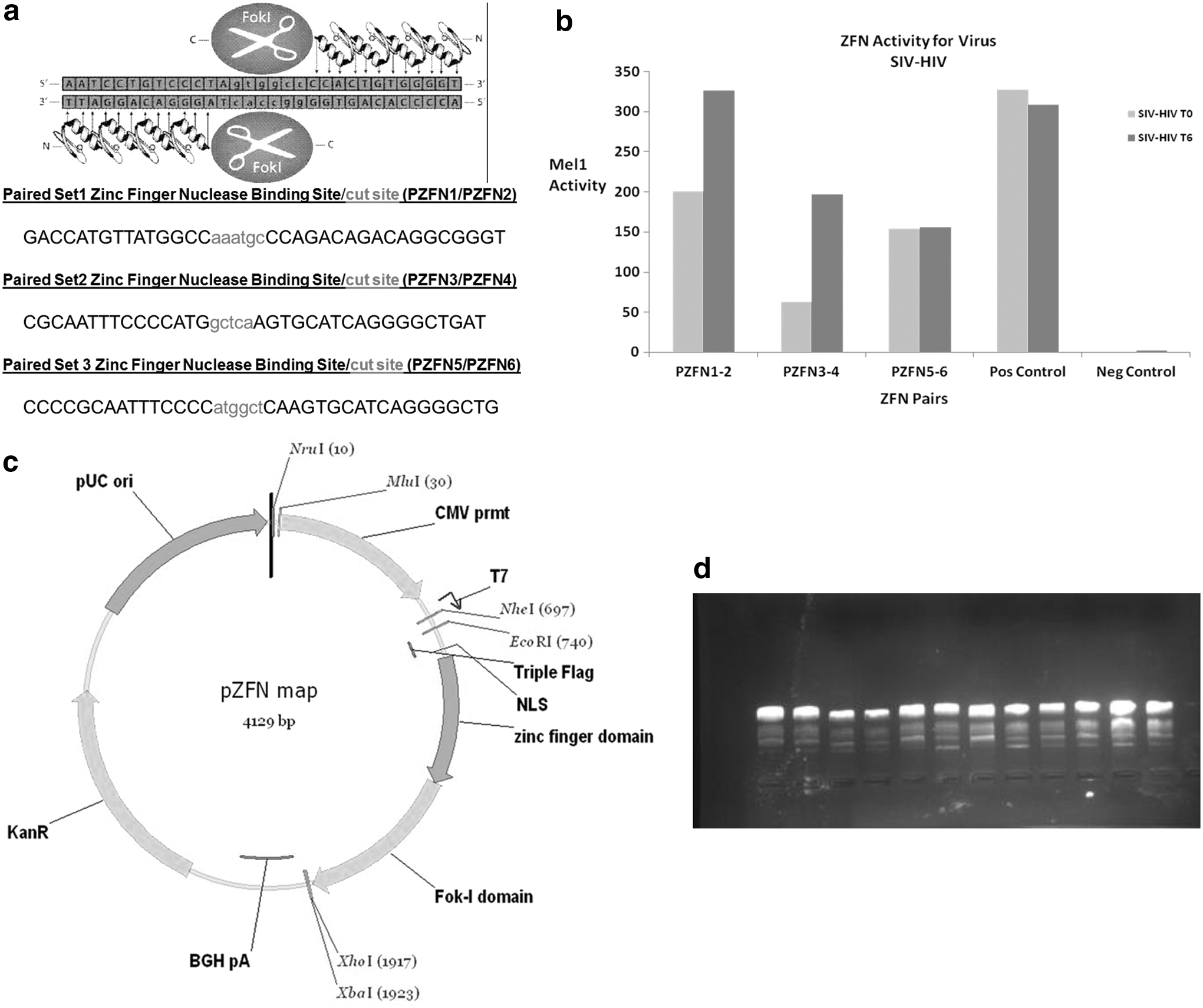
Construct and validation studies of HIV-1 pol gene targeting ZFN subclone in mammalian plasmids. This figures shows the construct and validation of ZFN in mammalian plasmids for their ability to target the HIV-1 pol gene using a MEL1 yeast assay in partnership with Sigma-Aldrich, GER.
RT-PCR, reverse transcriptase–polymerase chain reaction.
CRISPR/Cas-9 and pGS-U6-gRNA plasmids
A single pair of RNA genomic contigs corresponding to the CRISPR system targeting the 3,180 bp HIV-1 pol gene (denoted by order identify #s 462776-1 and 462776-2 or specifically HIV-1 pol gene _T1 and HIV-1 pol gene _T2) was selected from eight pairs cloned by PCR amplification. For each gRNA, all the potential off-target sequences across the human genome were identified and weighted according to sequence similarity. The gRNA sequences were ranked according to the risk level of off-target cleavage based on (1) sequence similarity and (2) location of off-target sites in intron or exon. Location of gRNA was considered. For knockout purpose, gRNA locates (1) at critical functional domain or (2) more to the 5′-end of coding DNA sequence is considered higher potential to knockout the critical functional domain or the encoded protein by inducing reading frameshift. On basis of these attributes, the top two gRNAs were recommended by our software and sequence validation and constructed into the proprietary gRNA backbone vector to form gRNA constructs for the CRISPR-cas9 genome editing system. Specifically, each arm of the pair was then separately subcloned into the 2,349 bp pGS-U6-gRNA plasmid core by CloneEZ (Fig. 2a, b, respectively). The pGS-U6-gRNA plasmid vectors basically comprise a pUC origin of replication located downstream of a U6 transcription promoter. In addition, kanamycin and zeocin resistance markers are integrated to allow for easy selection of transfected cells in culture. The quality of each plasmid vector was analyzed by sequencing and sequence alignment, PCR amplicon restriction fragment (XhoI and HindIII) gel electrophoresis, and DNA quality by NanoDrop. These are meant to be paired alongside the 7,037 bp pCMV-Cas or pCMV-Cas-D01A plasmids (Fig. 2c). Figure 2d and e shows the gRNA sequences of either CRISPR gRNA (T1, T2) and the gel electrophoresis of the entire pCMV-Cas or pCMV-Cas-D01A plasmids. 57 –59

Construct and validation of HIV-1 pol gene targeting CRISPR/Cas9. This figure details the construct and validation of CRISPR/Cas-9 guided RNA complexes targeting the same HIV-1 pol gene using PCR amplicon gel electrophoresis and sequencing in partnership with GenScript.
RGD adenoviral (Ad5) vector construct
To compare transduction and transfection efficiencies of mammalian plasmids and viral vectors, we subcloned the cDNA of each arm of the pair of HIV-1 pol gene targeting ZFN into rugged adenoviral vectors (cDNA loaned by Sigma-Aldrich, GER to Vector Biolabs). This process involved five stages, that is, cloning the gene of interest into p-cis shuttle vector, transfer of expression cassette into Ad vectors, first-generation, low-titer (HEK-293E cells), amplification and concentration, and finally, titration as summarized in Figure 3a. The issuing vector construct hosting a CMV promoter is shown in Figure 3b. Ad5 vectors were supplied at a concentration of 4.1 × 1,018 PFU/ml, and 2 × 1.0 ml was delivered. 60

Construct of RGD-Ad5 vectors carrying the HIV-1 pol gene targeting ZFN. This figures shows subcloning of the ZFN-consortium plasmids into an adenovirus type 5 rugged (RGD) vector in partnership with Vector Biolabs.
Cell lines, in vitro cell cultures, transductions, and luminescent or fluorescent marker assays
Cell lines
Through the ARRRP partnership, we obtained three continuous cell lines relevant toward HIV cure and/or vaccine research (order #20132587), namely TZM-bl (CAT #8129), ACH-2 (CAT #349), and J-LAT Full Length Clone 10.6 (CAT #9849).
In vitro culture, transduction, and assays
Briefly (1) “The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme, Inc.” TZM-bl, previously designated JC53-bl (clone 13) is a HeLa cell line.
61
–65
The parental cell line (JC.53) stably expresses large amounts of CD4 and CCR5. The TZM-bl cell line was generated from JC.53 cells by introducing separate integrated copies of the luciferase and β-galactosidase genes under control of the HIV-1 promoter. The TZM-bl cell line is highly sensitive to infection with diverse isolates of HIV-1. One milliliter per vial (4.2 × 106 cells/vial) provided to us in 50% fetal bovine serum (FBS), 40% DMEM, 10% dimethyl sulfoxide (DMSO) was thawed and seeded into culture flasks containing DMEM (90%), 10% FBS, 100 U of penicillin, and 0.1 mg/ml of streptomycin. Incubation in an Innova incubator was done at 37°C for 24–36 h. Cells grew adherently in a monolayer (Fig. 4a). This cell provided us with a model for studying the capability of both ZFN and CRISPR/Cas-9 to prevent primary HIV-1 infection. The TZM-bl indicator cell line enables simple and quantitative analysis of HIV using either β-gal or luciferase as a reporter. It is maximally sensitive to HIV infection by including DEAE-dextran in the infection medium. The β-gal and luciferase genes are also induced by HIV-2 infection. Following growth, cells were treated with ZFN plasmid, Ad-5 carrying ZFN, or CRISPR/Cas-9 plasmid, to protect them against incoming HIV-1. A transfection reagent was used. Effectively transfected cells were selected by growth on DMEM impregnated with the antibiotic zeocin. These cells were then challenged with HIV-1 clones isolated from supernatants of cultures of ACH-2, following which the cells were stained into a thin smear on slide and viewed under a fluorescent microscope. Treatment with either ZFN or CRISPR/Cas9 at 0.1, 0.2, 0.4, and 0.8 μl yielded increasingly reduced levels of luminescence visualized relative to untreated cells, following treatment with 1–2 μl of 0.34 mg of EnduRen
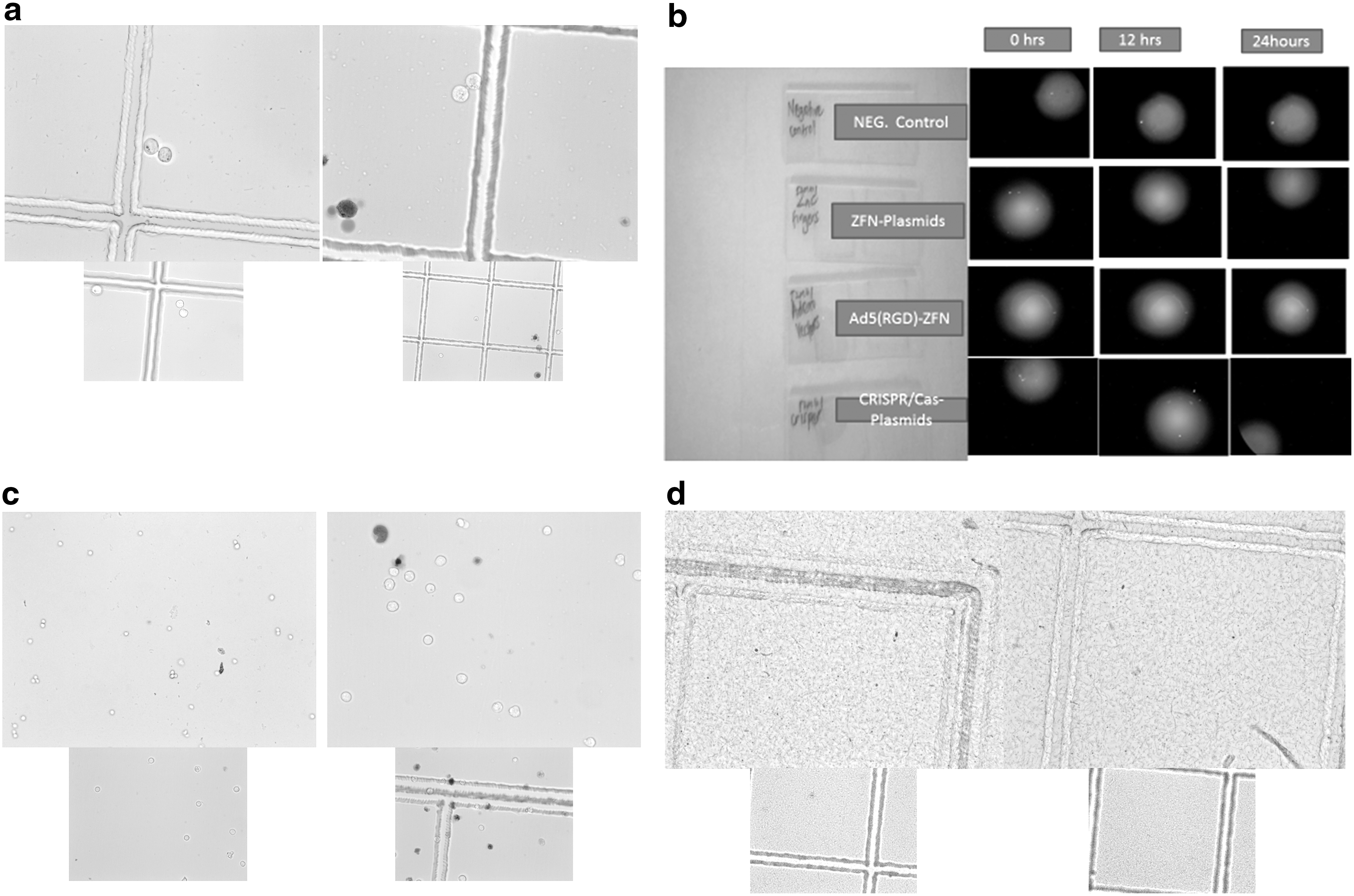
In vitro cultures, treatments, and/or challenge of cell lines with HIV-1 or activating agent. This figure shows in vitro cultures, treatments, and/or challenge of cell lines with HIV-1 or activating agent.
Target-mutagenesis validation by PCR amplicon digestion using a CEL1 assay in ACH-2 and J-LATs
To compare the efficiencies of HIV-1 pol gene target mutagenesis among ACH-2 and J-Lat cells treated with ZFN plasmids, RGD-Ad(5) ZFN vectors, or CRISPR/Cas9 plasmids, a Surveyor Mutation Kit from Integrated DNA Technologies (also known as the CEL1 assay Kit

Surveyor mutation of proviral HIV-1 pol gene targeting in ACH-2 and J-Lat. This figure details results of an SMA of proviral HIV-1 pol gene targeting in ACH-2 and J-Lat, revealing that Ad5(RGD) vectors enhanced ZFN splicing performance to equal or approximate that of CRISPR/Cas9 plasmids
qRT-PCR to examine extent of mutagenic viral infectiousness and reactivation
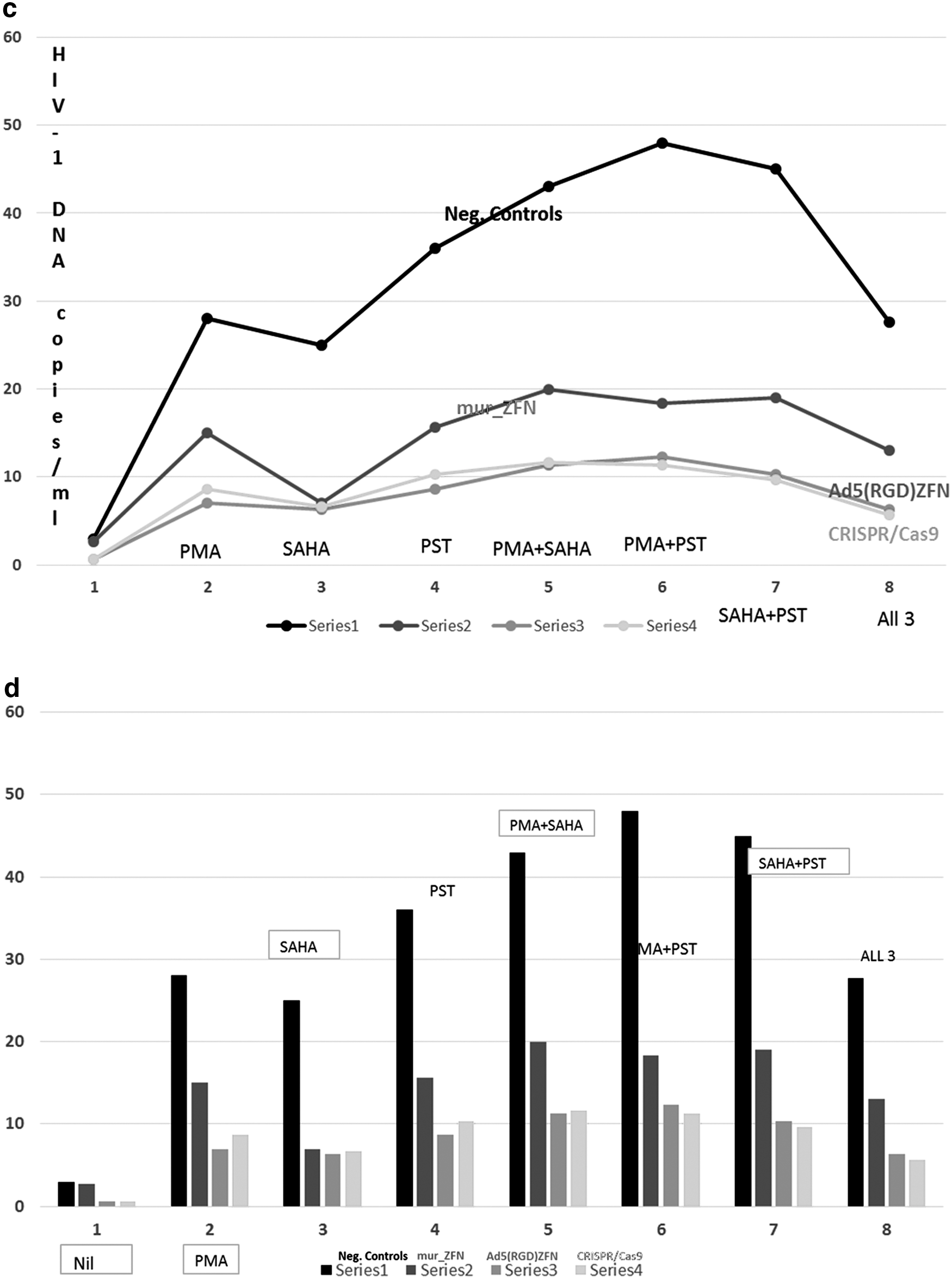
To quantitatively assay the abrogating effects of HIV-1 pol gene target mutagenesis on primary HIV-1 infection among TZM-bl cells or disabling the reactivation of integrated latent provirus among ACH-2 and J-Lat cell lines, we conducted qRT-PCR on effectively transfected cells (selected on basis of growth on zeocin) to determine the levels of expression of the pol gene transcript and viral copies/ml. With regard to abrogation of HIV-1 reactivation among ACH-2 and J-Lat cells, both Ad5(RGD)_pZFN and CRISPR/Cas9 achieved similar high performance relative to similar consortium plasmids (murCSTD_pZFN) (Fig. 6a, b vs. Fig. 6c, d, respectively). On a related note, both ZFN and CRISPR/Cas9 targeting the HIV-1 pol gene prevented primary HIV-1 infection of treated TZM-bl cells relative to untreated negative controls (10-fold and more) (Fig. 6e, f). In this respect, CRISPR/Cas9, however, seemed to abolish HIV-1 infectivity more than Ad5 (RGD) ZFN. The murCSTD_pZFN plasmids had the least impact to this effect. This was attributed to the enhancement of ZFN delivery by RGD-Ad5 relative to consortium plasmids (murCSTD_pZFN). These results are collaborated by the luminescence results seen among TZM-bl in context of halting primary HIV-1 infection by the same constructs. Imperatively, both ZFN and CRISPR/Cas9 targeted proviral HIV-1, although at varying degrees depending on their mode of delivery. Ad5 (RGD) vectors certainly improved ZFN delivery. A secondary outcome of these experiments is the revelation that combining the three epigenetic and nonepigenetic latency reversing agents did not perform better either one alone or just two. Theoretically, there thus appears to be an inhibitory effect of either latency reversing agent on one another when used in combination.

Quantitative reverse transcriptase assay of free HIV-1 viral loads among treated cell culture supernatants. This figure details results of qRT-PCR of free HIV-1 viral loads among treated cell culture supernatants.

Discussion
The continued lack of an effective vaccine and/or cure to HIV-1 remains a global public health challenge with drastic socioeconomic ramifications. 1,2,17 More recently, several ARE complexes have emerged as novel in vitro molecular entities for research and development (R & D) of an HIV-1 cure. These include native bacteria restriction enzymes (REases), ZFN, homing mega nucleases, transcription activator-like endonucleases, and clustered palindromic CRISPR/Cas9. In as much as the underlying mechanism of action for all these complexes is essentially similar—their effectiveness toward curing HIV has only been examined separately and using differing delivery vehicles. 40,52,54 –59,69 –73 If we are going to optimize a gene therapeutic cure (or prophylaxis) for eventual advancement to clinical trials in humans, more needs to be done to compare transduction and target-mutagenesis efficiencies of not only the various AREs presented to us but also the various options available for their delivery. Specifically, our team has proposed bone marrow infusion as a one-step gene therapeutic cure or treatment for HIV-1 using these AREs. To this effect, we compared the in vitro transduction and target-mutagenesis efficiency of HIV-1 pol gene targeting ZFN and CRISPR/Cas9 delivered as mammalian ZFN-consortium-plasmid, adenoviral (RGD-Ad5) vector, and CRISPR/Cas-9 plasmid among cultures of TZM-bl, ACH-2, and J-Lat cell lines toward an HIV cure (Tables 1 –3; and Figs. 1a–d, 2a–e, and 3a–b). Our data show for the first time ever that although CRISPR/Cas9 may possess inherent superior target-mutagenesis efficiency, the efficiency of ZFN (off-target toxicity withstanding) can be enhanced by altering delivery vehicle from consortium-plasmid to Ad5 (RGD) vectors. Moreover, we elucidate for the first time that combining three existing HIV-1 latency reversing agents—PST, PMA, and SAHA—did not offer better performance compared with either one or two, intimating a tertiary inhibitory effect (Table 4, and Figs. 4a–d, 5a, b, and 6a, b).
First, we constructed and validated constructs of ZFN in mammalian plasmids for their ability to target the HIV-1 pol gene using an MEL1 yeast assay in partnership with Sigma-Aldrich, GER (Tables 1 –3; and Fig. 1a–d). Then, we constructed and validated CRISPR/Cas9 guided RNA complexes targeting the same HIV-1 pol gene using PCR amplicon gel electrophoresis and sequencing in partnership with GenScript (Fig. 2a–e). Finally, we subcloned the ZFN-consortium plasmids into an adenovirus type 5 rugged (RGD) vector (Fig. 3a, b). Second, we cultured TZM-bl cells (that served as an in vitro challenge model for primary HIV-1 infection) and either ACH-2 or J-Lat clone 10.6 (as models of latent HIV-1 infection and reversal). Third, we either (1) transduced separate subcultures TZM-bl with the three study intervention constructs followed by challenge with primary HIV-1, measuring GFP-luminescence driven by HIV-1 LTR as the variable for examining prevention of primary HIV-1 (Fig. 4a, b) or (2) transduced separate subcultures of ACH-2 and/or J-Lat with the three study intervention constructs followed by activation with PST, PMA, or SAHA alone or in combinations of two or all the three (see Fig. 4c, d, respectively, for growth characteristics). Selection of effectively transduced cells was based on cultures in medium containing zeocin or kanamycin, since either resistance marker was integral in plasmid or vector constructs studied.
On one hand, with regard to abrogation of HIV-1 reactivation among ACH-2 and J-Lat cells, both Ad5(RGD)_pZFN and CRISPR/Cas9 achieved similar very high performance relative to similar consortium plasmids (murCSTD_pZFN) (see Fig. 6a, b vs. Fig. 6c, d, respectively). On a related note, both ZFN and CRISPR/Cas9 targeting the HIV-1 pol gene prevented primary HIV-1 infection of treated TZM-bl cells relative to untreated negative controls (10-fold and more) (Fig. 6e, f). Our fluorescence data support the view that Ad5 (RGD)_ZFN and CRISPR/Cas9 achieve equally higher transduction efficiencies relative to the murCSTD_pZFN plasmids among cultures of TZM-bl following addition of 1–2 μl of 0.34 mg of EnduRen Renilla luciferase substrate, relative to negative controls. Microcosmically, this explains why Ad5 (RGD) ZFN and CRISPR/Ca9 plasmids performed better than the murCSTZFN plasmids to this effect (Fig. 4b). The TZM-bl indicator cell line enables simple and quantitative analysis of HIV using either β-gal or luciferase as a reporter. On the other hand, SMA of proviral HIV-1 pol gene targeting in ACH-2 and J-Lat revealed that Ad5 (RGD) vectors enhanced ZFN splicing performance to equal or approximate that of CRISPR/Cas9 plasmids (Fig. 6a, b). The qRT-PCR assay of pol gene transcripts not only confirmed that either ARE construct effectively blocked HIV-1 challenge among TZM-bl but also supported the view that Ad5 (RGD) vectors enhanced the target-mutagenesis effectiveness of ZFN performance to approximate that of CRISPR/Cas9 (Fig. 6). Therefore, whereas several in vitro studies of ZFN and CRISPR/Ca9 targeting HIV-1 have been conducted previously, only our work simultaneously compares the two AREs alongside three plasmid or vector gene delivery vehicles. 54 –59,69 –73
In conclusion, whereas CRISPR/Cas9 may possess inherent superior target-mutagenesis efficiency; the efficiency of ZFN (off-target toxicity withstanding) can be enhanced by altering delivery vehicle from plasmid to Ad5 (RGD) vectors. Near-future research toward an HIV-1 gene therapy and/or prophylaxis must focus on enhancing transfection and transduction efficiencies of the available plasmid, viral vector, and/or other nanotechnologies for ARE delivery in vivo. Triple combinations of epigenetic and nonepigenetic HIV-1 latency reversing agents PST, PMA, and SAHA do not perform better than one or two agents, intimating inhibitory effects.
Footnotes
Acknowledgments
The authors appreciate Ms. Joanitta B. Birungi, who served as administrator for this project. Several administrative and laboratory scientists at Makerere University made this work possible indirectly. They specifically thank team members of the Immunology Laboratory in the College of Health Sciences (Samuel Kirimunda) and the Translational Laboratory of the Infectious Diseases Institute (Ms. Grace Turyasingura and Dr. Andrew Kambugu-IDI). Dr. Prosy Naluyima at MU-Walter-Reed Institute offered help on flow cytometry and fluorescent microscopy. Ms. Florence Achieng Oloo and Moses Aloo, who served as Program Officer for the GSK Trust in Science Africa Program, offered us indispensable oversight and guidance. The authors acknowledge the GSK Outreach Administrative and Legal teams, HIV DPU Biology team, and Genetics team, including Dr(s). Isro Gloger, Kevin Madauss, David Irlbeck, Rob Ferris, Charles Cox, Justin Rubio, and Beth Harris. This work was funded wholly by a GSK Trust in Science Africa Initiative Grant # COL100034581.
Author Disclosure Statement
All authors declare that there are no potential sources of financial conflicts of interests. M.W. is affiliated to Restrizymes Biotherapeutics (U) LTD that is researching the application of AREs as therapeutics and/or genomics vaccines against chronic viral infections in humans. M.W. has previously filed a related patent at the Uganda Registration Services Bureau (URSB) and African Regional Intellectual Property Organization (ARIPO: UG/P/2013/000009 “Zinc finger nucleases (ZFN) or any such future synthetic analogs and their use both as a gene (i) therapeutic cure and/or (ii) preventive vaccine, against persistent viral infections, specifically: proviral HIV-1, HSV-1 & 2, and high-risk HPVs”).