
Abstract
The clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas9 system has been used to excise the HIV-1 proviral genome from latently infected cells, potentially offering a cure for HIV-infected patients. Recent studies have shown that most published HIV-1 guide RNAs (gRNAs) do not account for the diverse viral quasispecies within or among patients, which continue to diversify with time even in long-term antiretroviral therapy (ART)-suppressed patients. Given this observation, proviral genomes were deep sequenced from 23 HIV-1-infected patients in the Drexel Medicine CNS AIDS Research and Eradication Study cohort at two different visits. Based on the spectrum of integrated proviral DNA polymorphisms observed, three gRNA design strategies were explored: based on the patient's own HIV-1 sequences (personalized), based on consensus sequences from a large sample of patients [broad-spectrum (BS)], or a combination of both approaches. Using a bioinformatic algorithm, the personalized gRNA design was predicted to cut 46 of 48 patient samples at 90% efficiency, whereas the top 4 BS gRNAs (BS4) were predicted to excise provirus from 44 of 48 patient samples with 90% efficiency. Using a mixed design with the top three BS gRNAs plus one personalized gRNA (BS3 + PS1) resulted in predicted excision of provirus from 45 of 48 patient samples with 90% efficiency. In summary, these studies used an algorithmic design strategy to identify potential BS gRNAs to target a spectrum of HIV-1 long teriminal repeat (LTR) quasispecies for use with a small HIV-1-infected population. This approach should advance CRISPR/Cas9 excision technology taking into account the extensive molecular heterogeneity of HIV-1 that persists in situ after prolonged ART.
Introduction
P
Current efforts to remove HIV-1 from the CD4+ memory T cell compartment primarily focus on a “shock-and-kill” strategy, in which drugs are used to induce reactivation of virus from latently infected cells. The intended outcome of this therapeutic approach centers on the elimination of latently infected cells after reactivation of HIV-1 transcription, replication, and infectious virus production because of cytopathic effects induced by the virus or by the host immune response. In principle, the latency reversal agents (LRAs) currently under evaluation include histone deacetylase inhibitors, protein kinase C agonists, farnesyl transferase inhibitors, and others as previously reviewed. 6,8 All of these experimental approaches involve varying degrees of activation of viral antigen production that is then targeted by the host immune system. However, in practice, some form of immune boosting will likely be required for this type of strategy to work optimally. 9 –11 A recent study has shown that the cytotoxic T lymphocyte (CTL) response depends on which LRA was used, indicating that not all LRAs will be able to induce a robust CTL response. 12 Additional studies involving reversal agent induced CTL suppression has been reported with different classes of LRAs, 13 having shown that a large proportion of the integrated proviruses in the latent reservoir are nonfunctional or unable to produce replication-competent virus under defined experimental circumstances at a given time point; the fraction of latently infected cells was shown to increase the longer the patient was suppressed with ART. 14,15 However, recent studies have indicated that some defective proviruses still have the ability to produce viral proteins, and this may be involved in shaping the overall CTL response, shifting its focus away from cells that have replication-competent proviral insertions. 16 Additional studies have suggested that only some types of CD4+ T cells infected with HIV-1 are being reactivated with the shock-and-kill approach. In this regard, viral outgrowth assays have shown that the proportion of CD4+ T cells reactivated from the resting CD4+ T cell HIV-1 reservoir was typically >100-fold lower than the number of infected cells detectable by quantitative polymerase chain reaction (PCR). 17 In addition, the efficacy of the “shock-and-kill” strategy with respect to eliminating or reducing the number of infected cells in the monocyte–macrophage lineage or within cells of the CNS remains unknown.
Given these observations, an alternative objective has been to find novel approaches that reduce the size of the latently infected cell population that do not require activation of HIV gene expression or reactivation of virus production. 18 This has largely entailed precise editing of the genome using one of the three main gene-editing systems. The zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are protein-based systems that can efficiently induce double-stranded DNA breaks (DSBs) but require in-depth design and modeling to ensure proper configuration of numerous tandem protein modules to recognize specific sequences in the genome. Unlike ZFNs and TALENSs, the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated Cas9 system is an RNA–protein-based gene-editing system, which has been widely implemented because of the ease of guide RNA (gRNA) design; implementation of specific gRNAs into a variety of CRISPR vector systems; and adaptability with respect to a large number of downstream applications. 19
The CRISPR/Cas9 system is being used throughout agricultural, biomedical, and basic research domains with ambition of this technology to be used in clinical trials. The two main components in the CRISPR/Cas9 system are the Cas9 endonuclease and the gRNA. The DNA sequence targeted by the CRISPR/Cas9 gRNA, also known as the protospacer, is complementary to the gRNA. This targeting event is facilitated by the recognition of a three-nucleotide protospacer adjacent motif (PAM) followed by a complementary sequence that stretches for ∼20 nucleotides immediately adjacent. 20 Once this binding reaction has been completed, Cas9 induces a DSB and the cellular DNA damage response repairs the break by nonhomologous end joining, thus inducing insertions and deletions (Indels).
The potential of CRISPR/Cas9 as a tool to eliminate the integrated HIV-1 provirus from latently infected cells has been demonstrated in vitro, in vivo, and more recently ex vivo. 21 –24 It has also been demonstrated to be capable of disrupting the CCR5 and/or CXCR4 coreceptors for viral entry. 25 –35 In addition to inducing Indel mutations around the target site, CRISPR/Cas9 has also been used to excise nearly the entire proviral genome and to activate latent genomes. 21 –59 The most promising results have demonstrated that HIV-1-specific gRNAs can target provirus excision from ex vivo patient samples. 23 In addition, the use of experimental animal models has demonstrated that CRISPR/Cas9 can be delivered to infected cells in the peripheral blood as well as the CNS and gut associated lymphoid tissue to induce Indels and excise provirus. 22,24 These experiments have produced ground-breaking results with respect to efforts to eliminate or “cure” an HIV-1-infected cell. However, in all these studies, there have always been varying amounts of residual provirus remaining in the infected cell population under investigation. We propose that this is, in part, because of the fact that the gRNAs used do not adequately account for the natural genetic diversity within the integrated HIV-1 proviral genomes within an infected patient, because viral quasispecies (vQS) continuously arise by mutation throughout the course of the disease even after prolonged ART. 60 –63
One major question in designing gRNAs for proviral excision is whether the vQS present within patients are homogeneous enough to use a small number of gRNAs directed against all proviral reservoirs remaining after successful HAART-mediated suppression? A recent study has sought to measure how genetic changes accumulate in the HIV-1 genomes of patients before and after HAART. 60 It was demonstrated that mutations continue to accumulate long after HAART-mediated suppression and can be clinically important. 60,63 A study of 36 HIV-1-infected patients enrolled in the Drexel Medicine CNS AIDS Research and Eradication Study (CARES) cohort found that—within the peripheral blood mononuclear cell (PBMC) compartment—the predominant long terminal repeat (LTR) sequence from integrated provirus exhibited decreased mutation rates after introduction of HAART, but genetic changes continued to accumulate, even after >6 years of therapy with a median of 10–20 unique mutations per year over the entire LTR region. 60 A similar study by Josefsson et al. examining 12 patients on long-term HAART reported that within CD4+ memory T cells and gut reservoirs, the gag-pol region of the genome mutated at a rate of 50–70 nucleotide changes per 10 kb per year before initiation of HAART, which was reduced to ∼1 nucleotide change per 10 kb per year during long-term HAART therapy. 64 The 10-fold difference in observed mutation rates after HAART-mediated suppression likely reflects the examination of distinct cell types and HIV-1 genomic regions within the two studies. As a single point mutation can disrupt CRISPR activity, it is important to tailor therapeutic molecules to individual patient vQS or otherwise ensure that gRNA design acts broadly across target sites despite genetic variation at any location across the HIV-1 proviral genomes. Understanding the importance of genetic variation for CRISPR/Cas9 design is further delineated by recent studies that show how variation in the human genome also needs to be considered for gRNAs with potential for therapeutic applications to minimize or avoid unwanted off-target introduction of DSBs. We have previously shown with eight peripheral blood samples from six HIV-1-infected patients that gRNAs tailored to individual patients could be used to target the detectable range of vQS present in each of the eight patient samples, as determined by next-generation sequencing. 61 In this study, longitudinally deep-sequencing data from 23 patients enrolled in the Drexel CARES cohort were used to determine whether broad-spectrum (BS) and personalized gRNA packages can be designed to target the HIV-1 vQS in patient-derived sequences.
Materials and Methods
Patient selection
Patients enrolled in the Drexel Medicine CARES cohort were recruited from the Partnership Comprehensive Care Practice of the Division of Infectious Disease and HIV Medicine in the Department of Medicine at Drexel University College of Medicine (Philadelphia, PA) and the Center for Clinical and Translational Medicine in the Institute for Molecular Medicine and Infectious Disease at the Drexel University College of Medicine. Patients in the Drexel Medicine CARES cohort were recruited under protocol 1201000748 (B.W., Principal Investigator), which adheres to the ethical standards of the Declaration of Helsinki (1964, amended most recently in 2008), which was developed by the World Medical Association as previously described. 65 All patients provided written consent upon enrollment. For this study, 23 patients were selected that used the following selection criteria: (i) patient samples were available for analysis from three or more patient visits, (ii) patients were currently on HAART, (iii) patients whose last recorded CD4+ T cell count was ≥350 cells/μL, (iv) patients whose last recorded viral load was ≤50 copies/mL, (v) patients who had a visit during the 2016 calendar year, (vi) patients had a normal neurocognitive assessment, 66 (vii) patients were hepatitis C virus-negative at all visits, and (viii) patients who were found to test negative for drugs of abuse at their most recent visit; however, some individuals do have a history of drug abuse (Table 1). Each patient then had samples analyzed from two time points, with two patients having three longitudinal samples.
ART, antiretroviral therapy; LTR, long terminal repeat.
Deep-sequencing methodology
Amplification of patient HIV-1 LTRs and deep sequencing were performed as previously described.
60,63,65,67
In brief, patient PBMCs were isolated by Ficoll gradient centrifugation. Genomic DNA was isolated and a HIV-1 LTR-specific two-round nested PCR was performed. The amplified LTRs were then purified and tagmented using the Nextera XT Library Prep procedure with the Nextera XT Index procedure v2 to produce the sequencing libraries. Sequencing was performed using the Illumina NextSeq 500 instrument in single-ended mode using a 150 Mid Output v2 sequencing procedure, as previously described by the manufacturer (Illumina). This produced ∼10 million reads per sample, providing the ability to query diversity of the HIV-1 LTR within PBMCs. All samples were sequenced to an average depth of at least 1,000 × across the LTR (positions 36–586) (Supplementary Fig. S1A; Supplementary Data are available online at
gRNA design
From the positional frequency data of the vQS, gRNAs were designed in a two-stage process. First, the consensus sequence of the 21 nt 5′ of all GG dinucleotides in the reference sequence (on both strands) was extracted from each patient sample to produce a candidate list of gRNAs. The consensus sequence was selected as the gRNA, as opposed to the most common 21-mer, because the consensus sequence will have the lowest number of mismatches when compared across all reads; this is not always true of the most common 21-mer. Second, the methodology, previously introduced in Dampier et al., was expanded to work with next generation sequencing (NGS) data. 62 This was accomplished by utilizing the Massachusetts Institute of Technology (MIT) penalty function 70 (Fig. 1) to find gRNAs with a BS activity. An MIT penalty score of 1 indicates a perfect match and assumes 100% predicted cleavage (Fig. 1C, D). To predict what fraction of a patient's vQS would be cleaved by a particular gRNA candidate, a two-step process was used. First, all aligned reads overlapping the gRNA target site were extracted from the alignment. Second, a sliding window was used on each read to determine the likelihood that a query gRNA would be predicted to cleave that patient-derived target sequence even in the presence of small Indels within the read. This result was then averaged across all reads that cover the target region. Base quality scores were not considered in this step. It is mathematically more likely that a sequencing error would decrease the binding score and as such these values underestimate the likely predicted cleavage efficiency. The predicted cleavage of a gRNA for a particular patient-derived sequence was determined up to the limit of detection, which is determined by the sequencing depth across the target position. Multiplexes of gRNAs were considered as independent observations and aggregated by calculating the likelihood that any gRNA would be predicted to cut the target sequence. The final list of gRNAs was then screened against the human genome using a BLAST query against HG19. Considering a gRNA has 20 nucleotide positions, the gRNAs that had 17 or more nucleotide matches at any site in the human genome were excluded, as they have a high chance of binding to the human genome. The gRNAs that had 16 or less matches to the human genome were kept, as these have minimal if any chance of binding to the human genome and causing off-target effect.

gRNAs were designed against patient-derived HIV-1 quasispecies. The position-specific penalties associated with mismatches within the protospacer used the MIT penalty score
gRNA selection
The final candidate list of gRNAs was sorted by the total percentage of observed vQS that would be predicted to be cleaved within that particular sample (personalized) or across all samples (BS) in the study. With the assumption that the final delivery vector could accommodate a multiplexed delivery of four gRNAs (maximum currently able to be placed into a lentiviral vector system), three types of gRNA packages were designed: the top four gRNAs with the greatest total cleavage potential within each individual (personalized; PS1–PS4) or across all samples (BS; BS4), as well as the top three BS gRNAs plus one personalized gRNA for each individual (3 + 1 package; BS3 + PS1). The personalized gRNA was selected from the earlier of the two visits from each patient and the most effective patient-specific gRNA was added to the collection.
Results
A personalized gRNA package was effective at vQS recognition over an extended period of time
Many of the gRNAs specific to HIV-1 have been designed from a single input sequence or from a small number of laboratory strains. In this study and unique when compared with others, anti-HIV-1 gRNAs specific for the HIV-1 LTR were designed based on thousands of patient-derived HIV-1 sequences. To design HIV-1-specific gRNAs, 23 patients with at least two visits were selected for deep sequencing of the HIV-1 LTR from the Drexel Medicine CARES cohort (described in the section “Materials and Methods” and Table 1). PBMC isolation from peripheral blood samples and HIV-1-specific LTR PCR for NGS was performed as previously described. 63,65,67 Using the algorithm already described, gRNAs were designed for each of the 48 samples to optimize cleavage efficiency in the face of LTR genetic diversity. The algorithm identified regions of high conservation within the LTR and ensured they matched the seed region (positions 15–20) within the gRNA (positions within the protospacer associated with high penalty), whereas LTR regions with high diversity were accommodated with protospacer positions with little or no associated penalty (Fig. 1). Of the 48 samples examined, 45 were cleavable to the limit of detection with 3–8 gRNAs (Fig. 2A). This implies that the vector design needs to accommodate at least four configurable gRNAs, which has been shown for a lentiviral vector previously, and more recently using an adeno-associated virus (AAV) vector. 24,71 When the top 4 gRNAs were individually used for each sample to calculate the predicted percentage cutting against its own sample (personalized), 43 of the 48 were predicted to be cleaved to 99% efficiency and all 48 samples would be cleaved to 90% efficiency (Fig. 2B).
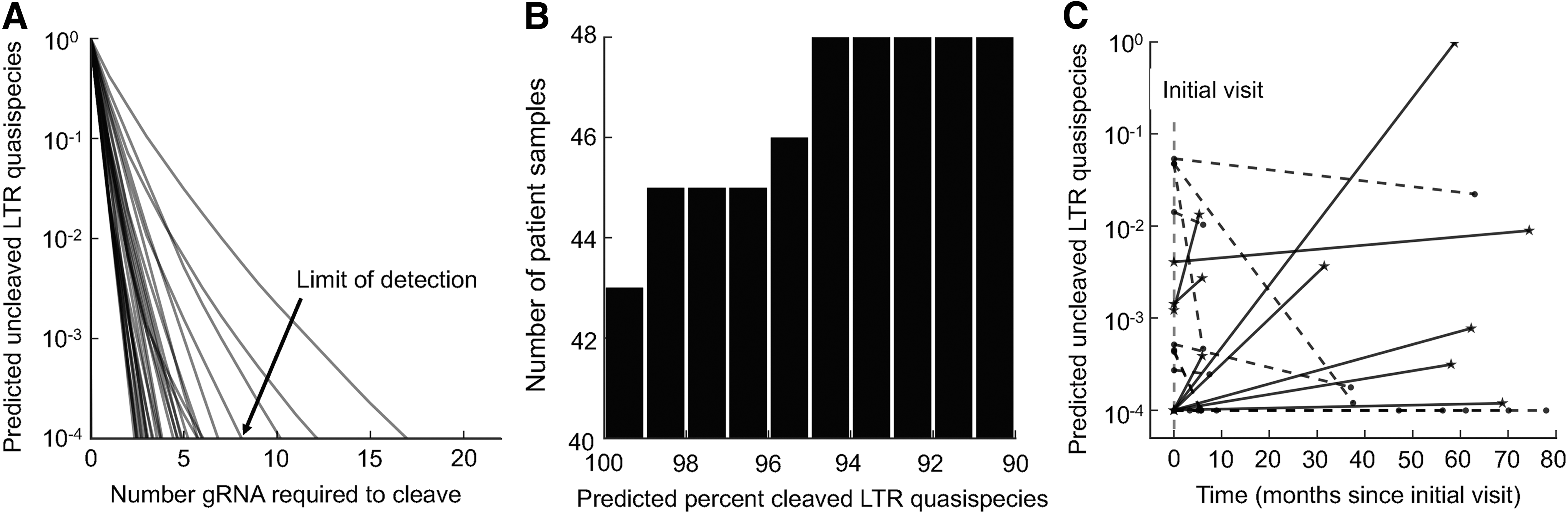
Personalized gRNAs can target the quasispecies with longitudinal efficacy.
If it is necessary to devise a personalized set of gRNAs to excise the integrated provirus, it is obvious that there will be a delay between sample isolation, sequencing of the vQS, design of the gRNAs, and construction of the therapeutic for delivery. At present, the length of this time frame is not known. However, as indicated in our previous study, 61 designing gRNAs for one visit may not be as effective on subsequent visits because of continued evolution of proviral sequences. However, that statement was made with only a few longitudinal samples evaluated. Given that these 48 samples were from 23 patients with two or three visits each, this larger data set was used to more accurately address this question. Utilizing the current data set, there was not a consistent pattern with respect to the effectiveness of personalized therapies as time progresses (Fig. 2C). In a total of 16 of the visit pairs, a decreased effectiveness was observed while 12 either maintained or improved in their effectiveness regarding predicted cleavage efficiency. This implies that there may be a time limit between the sampling and delivery of a gRNA package for some patients. This result also reconfirms that some patients on HAART still have ongoing, although low level, genetic variation that could have a functional impact on the efficacy of therapy. However, the level of genetic variation was also shown to be patient specific, with some personalized gRNAs remaining effective at cleaving the vQS even up to 70 months later (Fig. 2C). Therefore, continued NGS analysis of longitudinal samples is important to designing gRNAs to tackle the entire vQS within and among infected individuals.
A BS gRNA package can be designed to eliminate the vast majority of vQS in a given patient
When the LTRs from all 48 samples were pooled together, there were five gRNAs that had a cutting fraction >10%, with the optimal one being able to cut ∼90% of the quasispecies observed in all samples (Fig. 3A). The top four gRNAs were predicted to cut the majority of patient-derived sequence in all samples and were used in further analysis as the BS package (BS4). The gRNA sequences are included in Supplementary Table S1). One of the major questions in gRNA design is how well gRNAs designed from one set of data can potentially cut proviral DNA from nonevaluated specimens. We utilized the methodology previously described in Dampier et al. to evaluate how well this package of gRNAs would function across the patient-derived sequences from the Los Alamos National Laboratory Database (

A package of four gRNAs has potential BS cutting efficiency.
Owing to practical limitations in delivering a purely personalized treatment and the high success rate of the BS package, a hybrid method (utilizing some consensus gRNAs and some personalized gRNAs) was used to compare with a BS (BS4)-only approach. In theory, this process could involve the production, validation, and experimental utilization of a package of three BS gRNAs along with a more extensive library of personalized gRNAs. The design process was examined by combining the top three BS gRNAs (BS3) with one personalized gRNA (PS1) to see whether this approach would increase the efficacy of the package of gRNAs (Fig. 4). The targets of BS3 and PS1 anti-HIV-1 gRNAs in each patient are given in Supplementary Table S1. These analyses demonstrated that the BS3 + PS1 package produced comparable results to those obtained utilizing the personalized gRNA or BS4 at 90% efficiency (45 patient samples predicted to cut with BS3 + PS1 vs. 46 with personalized, vs. 44 with BS4). However, at higher efficiency cutoff of 99%, the BS4 package underperforms personalized therapy (Table 2). The personalized gRNAs are not clustered in any particular part of the LTR as indicated by the large variation in their optimal targeting positions (Fig. 4).

Combining three BS gRNAs with a single personalized gRNA (BS3 + PS1) is predicted to cleave the majority of the patient quasispecies. On the left, the positions of each gRNA for each patient are shown with the shade indicating the weighted average of the quasispecies that was predicted to be cleaved. On the right, the combined weighted cleavage fraction was calculated for each patient assuming each position was independent.
BS, broad-spectrum.
Discussion
When considering gRNA design for HIV-1 excision therapy, one of the most important concerns is the extent that the therapy will be limited by the variation in the HIV-1 genome over time because of both the high mutation rate of the HIV-1 reverse transcriptase 72 and recombination events. 73 This rapid rate of mutation and a host of selective pressures ultimately lead to a swarm of similar, yet distinct, HIV-1 genomes within a given patient. This study has shown that it is possible to select a set of anti-HIV-1 LTR gRNAs that are expected to cleave all or nearly all vQS within an individual by taking advantage of the inherent promiscuity of the CRISPR/Cas9 system. We have demonstrated the use of patient-derived sequence information to inform gRNA design that can enhance CRISPR/Cas9 gRNA specificity rather than using a single input sequence to try and capture a diverse population. Furthermore, this study shows that a small package of gRNAs can be selected such that they will likely cleave a majority of the vQS within and across the patients tested.
To our knowledge, this is the first study to have designed HIV-1-specific gRNAs that are predicted to bind to a spectrum of vQS (within the limit of detection) for a vast majority of patients tested. Although current HIV-1-specific gRNAs are effective in a number of assays performed, 2,21 –30,32 –45,47 –59 they were not screened to test patient-derived sequences and indeed were sometimes specifically designed using the genomes of laboratory strains in an attempt to inhibit the expression of a particular HIV-1 gene, the activity of the viral promoter (LTR), or to abrogate viral replication. The gRNAs that are reported herein (Supplementary Table S1) are ideal for testing in a number of different therapeutic avenues for three reasons: (i) they can recognize all vQS within an infected patient to a given limit of detection, (ii) they are likely to prevent or dramatically delay virus escape due to being multiplexed and targeting conserved regions in a patient-specific manner, and (iii) could be easily incorporated into CRISPR/Cas9-specific lentiviral vectors or AAV that can accommodate up to four gRNAs.
There has been extensive research into in silico methods to predict the likelihood of CRISPR/Cas9 cleavage of particular gRNA:target pairs. A position-specific scoring matrix of CRISPR/Cas9 has been developed to estimate the likelihood of off-target effects from a given gRNA using systematically mismatching gRNA variants on the same target. 70 The promiscuous nature of gRNA targeting has been one of the primary concerns with regard to therapeutic gene-editing technology. A more recent study further expands the gRNA matrix to 12 × 20 possible substitutions as well as 64 possible PAMs. 74 In addition, these gRNA search algorithms were developed to improve off-target prediction by incorporating scoring matrices and evidence-based rules as previously reviewed. 75,76 These algorithms were incorporated into the pipeline presented here.
Along with the challenge of designing the gRNAs necessary for the excision of all integrated proviruses, the other major challenge will be delivery of the gRNAs. One way that has been proposed is the use of lentiviral and AAV vectors. The lentiviral delivery system would allow for the infection of similar cell types as HIV naturally infects, potentially limiting the delivery of therapy to uninfected target cells. However, the efficiency of vectors with respect to delivery to latently or persistently infected cells may be complicated by both the decreased infection rates of these types of cells, which have been previously infected by HIV-1 and show a more resistant phenotype to secondary HIV-1 infection. The lentiviral delivery system is also limited in the number of gRNAs that can be packaged into a single vector construct. Spreading the gRNAs across many vectors may result in a cell not receiving the gRNAs required for proper targeting. The nature of this limit has yet to be fully defined. Finally, even if the previous hurdles are overcome, an ideal therapy needs to deliver the gRNAs to tissue resident cells that are present at low numbers that may require lentiviral migration across vascular barriers.
The CRISPR/Cas9 system has been shown in vitro, in vivo, and ex vivo as a novel and promising method for treating and possibly curing HIV infection. 21 –24 In this analysis, we have demonstrated that the proposed gRNA design pipeline can lead to the production of anti-HIV-1 gRNAs that can span multiple viral genotypes as well as being patient specific. The BS4 or BS3 + PS1 strategy in silico clearly surpasses current gRNAs with respect to cleaving patient-derived HIV sequences that are detectable by NGS across relatively large number of HIV-1-infected patients. With an increased number of longitudinal patient-derived PBMC samples that have been subjected to NGS, this gRNA design pipeline has been shown to be capable of designing optimal gRNAs to target a broader spectrum of patients, from multiple cohorts. Future studies will be needed to examine the potential of gRNAs to target multiple HIV subtypes, beyond subtype B that is common in North America and Europe, to hopefully achieve a functional and/or sterilizing HIV cure.
Sequence Data
Final sequences have been submitted to the SRA and all samples were linked under Bioproject ID PRJNA309974.
Footnotes
Acknowledgments
These studies were funded, in part, by the Public Health Service, National Institutes of Health, through grants from the National Institute of Mental Health (NIMH) R01 MH110360 [Contact Principal Investigator (PI), B.W.], the NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (K.K., PI; B.W., PI of the Drexel subcontract involving the Clinical and Translational Research Support Core) and under the Ruth L. Kirschstein National Research Service Award T32 MH079785 (Dr. B.W., PI of the Drexel University College of Medicine component and Dr. Olimpia Meucci as Co-Director). All sequencing was performed at the Center for Genomics Sciences in the Drexel University College of Medicine Institute for Molecular Medicine and Infectious Disease. The contents of the article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author Contributions
W.D., M.R.N., and B.W. conceptualized the article. W.D., M.D., and M.R.N. analyzed the data and wrote the first draft of the article. W.D., N.T.S., M.R.N., V.P., C.-H.C., A.G.A., G.D.E., J.C.M., and B.W. contributed to writing and made critical revisions of the article. Z.S. and J.M.J. aided in recruiting patients. M.D., W.Z., K.K., S.P., and J.W.W. contributed to patient sample preparation and experimentation. All authors reviewed and approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.