
Abstract
Currently, complex HIV-1 recombinations among the B′, C, and CRF01_AE genotypes have frequently arisen in Yunnan, China. A novel HIV-1 complex circulating recombinant form (CRF) consisting of B′, C, and CRF01_AE (CRF96_cpx) was recently characterized from three epidemiologically unlinked individuals. Two strains of them were isolated from the injecting drug users in this study, the remaining one strain (JL. RL01) was obtained from a previous report in Jilin province. Phylogenetic analysis based on near full-length genome revealed that CRF96_cpx formed a distinct monophyletic cluster supported by a high bootstrap value of 100%, distantly related to all known HIV-1 subtypes/CRFs. CRF96_cpx had a CRF01_AE backbone with three subtype B′ and C segments inserted, respectively, in the gag and pol region. Furthermore, subregion tree analysis showed that CRF01_AE backbone and subtype B segment inserted originated from a Thai-CRF01_AE lineage, whereas subtype C fragment inserted was from an India C lineage. They are different from previously documented CRF01_AE/B/C forms in its distinct backbone, inserted fragment size, and breakpoints. This highlighted the importance of continual monitoring of genetic diversity and complexity of HIV-1 strains in Yunnan, China.
HIV-1
China has been seriously affected by HIV/AIDS; the prevalence of HIV infection is especially high in western Yunnan, which is located in southwestern China, borders the opium-producing “Golden Triangle” regions composed of Myanmar, Laos, Thailand, and Vietnam. HIV-1 transmission is closely associated with illegal drug trafficking. Yunnan is regarded as the epicenter of the HIV-1 epidemic in China. In addition, wide cocirculation of subtypes B′, C, and CRF01_AE among high-risk individuals provides the opportunity for the generation of various novel CRFs. Numerous newly identified CRFs of HIV-1, such as CRF07_BC, 2 CRF08_BC, 2 CRF57_BC, 3 CRF62_BC, 4 CRF64_BC, 5 CRF65_cpx, 6 CRF78_cpx, 7 CRF86_BC, 8 CRF87_cpx, 9 and CRF88_BC, 9 have been reported in Yunnan over the past 20 years. In recent years, complex HIV-1 recombination among the B′, C, and CRF01_AE genotypes has frequently arisen in this region. A recent study showed that a novel HIV-1 second-generation recombinant form originated from CRF01_AE, and CRF08_BC was identified here. 10 In this study, we characterized a new HIV-1 CRF, designated CRF96_cpx, on the basis of near full-length genome (NFLG) analysis, from the HIV-1 strains isolated from two injecting drug users in Baoshan prefecture, Yunnan, and an additional one strain was reported in a previous study in Jilin province. There was no epidemiologic link among these three individuals.
Plasma sample was collected from two HIV-positive patients (13YNBS54IDU and 13YNBS66IDU) who were intravenous drug users from Baoshan prefecture, Yunnan. The remaining one strain (JL. RL01) obtained from a previous report in Jilin province exhibited a high degree of genetic similarity with the two strains in this study through BLAST analysis. Basic epidemiological information is given in Table 1. The study was approved by the Kunming University of Science and Technology Ethics Committee. All participants supplied written informed consent for specimen collection and subsequent analyses.
The NFLG amplification and sequencing were performed as previously described. 11 In brief, RNA was isolated from 280 μl plasma using the Virus RNA Mini Kit (Tiangen) according to the procedure described in the manual. Then the near full-length HIV-1 genome was amplified separately using reverse transcription-nested polymerase chain reaction according to the method described in previous reports. The generated products were analyzed by agarose gel electrophoresis, and the positive PCR samples were purified using the PCR Product Gel Extraction Kit (Tiangen) and were then sequenced by Invitrogen Co (Guangzhou, China).
All of the overlapped subgenomic DNA sequences were successfully obtained from the two subjects. Then the sequenced fragments were edited and assembled into contiguous sequences using ContigExpress software. The sequencing data were aligned against the HIV-1 sequence database using NCBI BLAST search and manually edited with Bioedit 7.2.1 with reference to HXB2 to ensure accurate codon alignment. Phylogenetic tree analyses were carried out using the neighbor-joining method based on the Kimura 2-parameter model with 1,000 bootstrap replicates and a transition-transversion ratio of 2.0 applied in MEGA 6.02. The reference sequences relevant to HIV-1 epidemics in Asia were downloaded from the Los Alamos National Laboratory HIV sequence database. Recombination breakpoints were determined using Recombinant Identification Program included in the Los Alamos HIV database, and were further confirmed using SimPlot 3.5.1 software to perform bootscanning and informative-site analyses. Based on the information generated from Simplot, the structure of the new HIV-1 recombinant forms (01AE/B/C) was elucidated using the Recombinant HIV-1 Drawing Tool. To confirm the subtypes of mosaic fragments and analyze the origin of each segment, phylogenetic subregion tree analysis was conducted by the already mentioned method.
The NFLG sequences from the two subjects were 8,798 and 8,778 bp in size for strains 13YNBS54IDU and 13YNBS66IDU, respectively, spanning from the most of gag gene to part of three long terminal repeats corresponding to the location 790–9637 of HXB2 strain. Phylogenetic analysis was carried out based on the two sequences together with JL. RL01. The results showed that these three sequences formed a distinct monophyletic cluster supported by a high bootstrap value of 100%, distantly related to all known HIV-1 subtypes/CRFs (Fig. 1). Furthermore, both RIP and boot scanning analyses revealed that the three NFLGs share the same identical mosaic structures composed of CRF01_AE, B, and C, with three short subtypes B and C segments, respectively, inserted into the gag and pol regions of the CRF01_AE backbone. A total of eight recombinant breakpoints corresponding to HBX2 were determined using the informative sites analysis. The genomic map charted by the Map-Draw Tool available at the Los Alamos HIV sequence database is shown in Figure 2.

Phylogenetic analyses of the near full-length genome nucleotide sequence of CRF91_cpx. The representing different HIV-1 group M reference sequences were used to construct the neighbor-joining phylogenetic tree. The sequences of CRF91_cpx (13YNBS54IDU, 13YNBS66IDU, and JL.RL01) are marked in •. The stability of the nodes was assessed by bootstrap analysis with 1,000 replications, and only bootstrap values 100 are shown at the corresponding node. The scale bar represents 5% genetic distance.
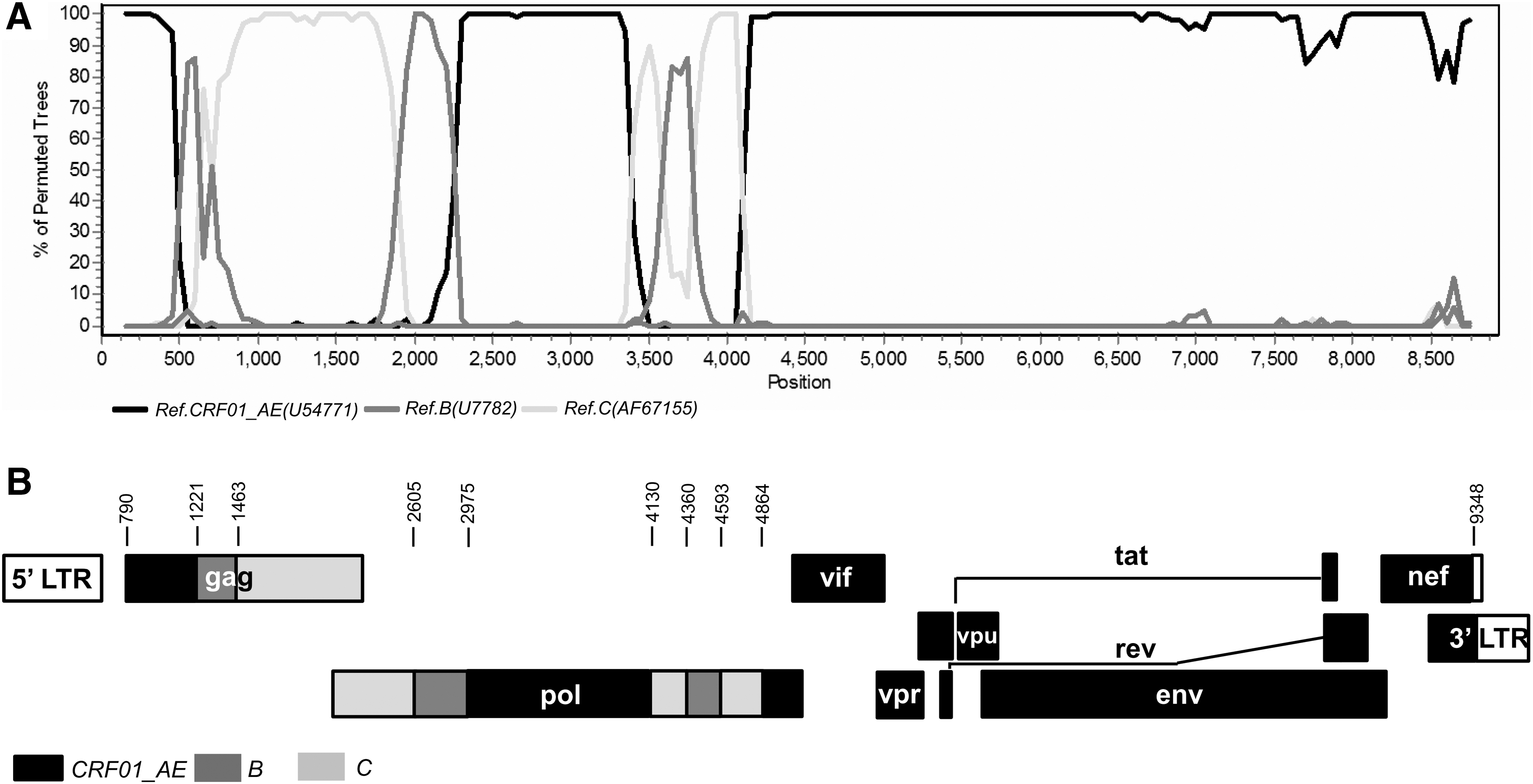
Recombination breakpoint analyses of CRF91_cpx.
Subregion tree analyses further confirmed the breakpoints of the three NFLG sequences as follows: I (790–1220 nt) CRF01_AE, II (1221–1462 nt) B, III (1463–2604 nt) C, IV (2605–2974 nt) B, V (2975–4129 nt) CRF01_AE, VI (4130–4359 nt) C, VII (4360–4592 nt) B, VIII (4593–4863 nt) C, and IX (4864–9637 nt) CRF01_AE, using HXB2 as a reference. Recombinant structures of these strains were distinct from the any known CRFs reported to date, and the strains were isolated from three HIV-1 infected patients without obvious epidemiological linkage. Therefore, these new recombinants are now designated CRF96_cpx. Moreover, subregion tree analyses also revealed that the backbone fragment of CRF01_AE (regions I, V, and IX) originated from Thailand CRF01_AE, the inserted three segments of subtype B (regions II, IV, and VII) were most likely from the Thailand and China B lineage (TH/CN), and the remaining three fragments of subtype C (regions III, VI, and VIII) originated from the India and China C lineage (IN/CN) (Fig. 3).
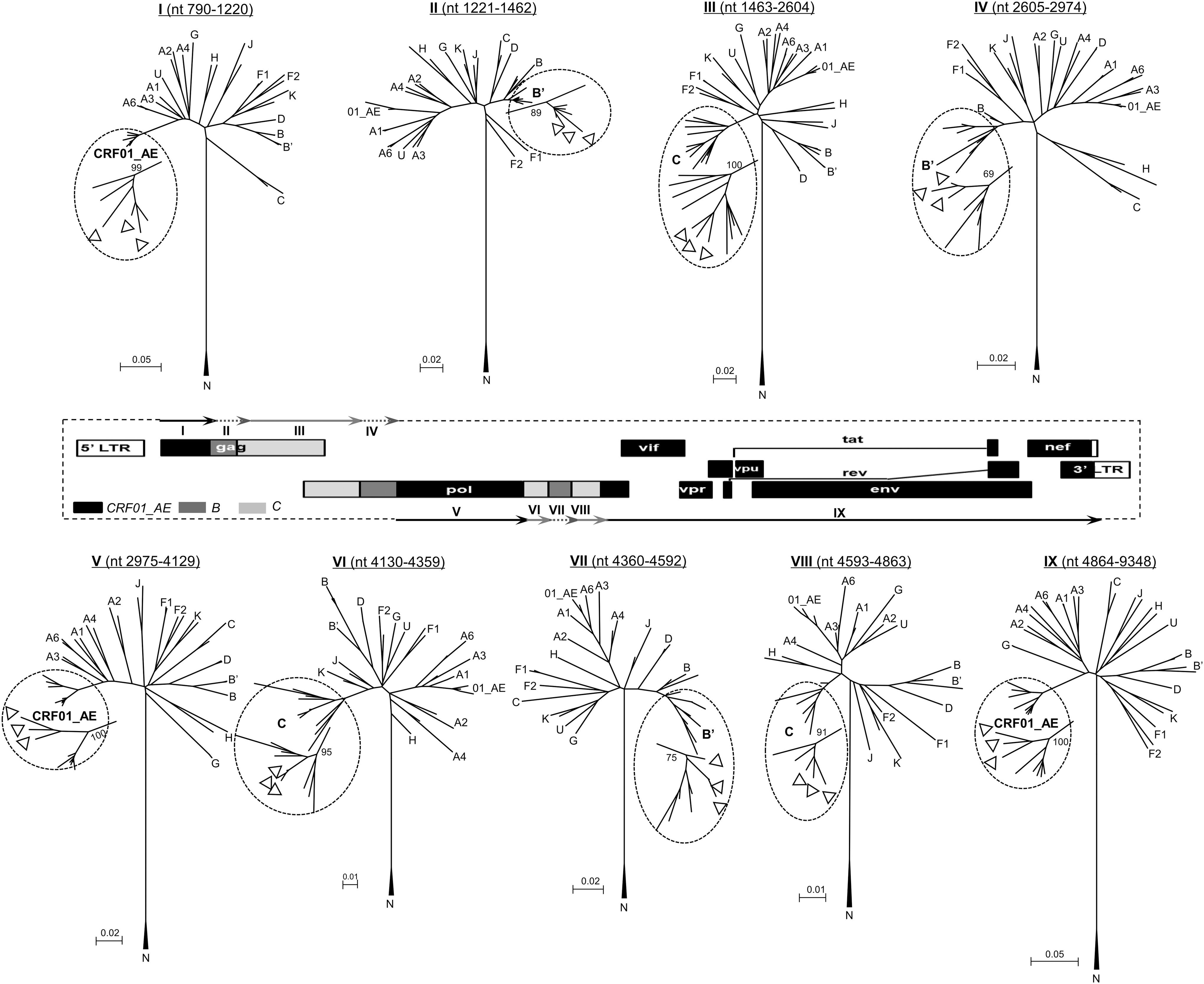
Subregion tree analyses of the CRF91_cpx genome. The phylogenetic trees of the nine mosaic fragments identified by bootscan analysis were constructed using the neighbor-joining method based on the K-2 model in MEGA. The reliability of tree branches was evaluated by 1,000 bootstrap replicates, and bootstrap values >0.7 were considered stable. The sequences of CRF91_cpx isolates are marked in △.
In this study, we describe a novel HIV-1 CRF consisting of CRF01_AE, B′, and C (named CRF96_cpx). This CRF had a CRF01_AE backbone with three subtypes B′ and C segments, respectively, in the gag and pol regions. The first two BC breakpoints (regions II, III, and IV) are similar to CRF08_BC form; it is possible that a CRF08_BC virus was involved in the history of this new form. Therefore, further molecular epidemiological surveys are essential to monitor the geographic origin and evolution history of CRF96_cpx. Moreover, to our knowledge, this is the fourth complex CRF identified in China. To date, a total of six complex CRFs involving CRF65_cpx, CRF77_cpx, CRF78_cpx, CRF82_cpx, CRF83_cpx, and CRF87_cpx have been identified in Asia. From the region distribution, CRF65_cpx and CRF87_cpx are documented in the Dehong prefecture. 6,9 The CRF96_cpx and our previously reported CRF78_cpx were found in Baoshan prefecture. 7 Moreover, CRF82_cpx and CRF83_cpx were reported in Myanmar. 12 Dehong and Baoshan are located in the western region of the Yunnan province and share its border with Myanmar. Taken together, our results again demonstrated that the high frequency of multiple recombinant events between B′, C, and CRF01_AE was constantly occurring in the areas on the China–Myanmar border, resulting in the formation of complex and diverse HIV-1 variants. Meanwhile, the evolving molecular epidemiological profile of HIV-1 poses a challenge for antiretroviral therapy. Therefore, it is necessary to strengthen the prevention and control measures for HIV-1 infection in this region.
Sequences Data
The NLFG sequences of isolated 13YNBS54IDU and 13YNBS66IDU have been deposited in GenBank under accession numbers MG518476 and MG518477.
Footnotes
Acknowledgments
This study was supported financially by National Natural Science Foundation of China (no. 81360247) and funded by New Product Development Projects of Yunnan Province (2016BC003 and 2016BC005), partially supported by Yunnan provincial innovation team project (2015HC030).
Author Disclosure Statement
No competing financial interests exist.