
Abstract
HIV-1 subtype B virus is the most prevalent subtype in Puerto Rico (PR), accounting for about 90% of infection in the island. Recently, other subtypes and circulating recombinant forms (CRFs), including F(12_BF), A (01_BF), and CRF-39 BF-like, have been identified. The purpose of this study is to assess the distribution of drug resistance mutations and subtypes in PR. A total of 846 nucleotide sequences from the period comprising 2013 through 2017 were obtained from our “HIV Genotyping” test file. Phylogenetic and molecular epidemiology analyses were performed to evaluate the evolutionary dynamics and prevalence of drug resistance mutations. According to our results, we detected a decrease in the prevalence of protease inhibitor, nucleoside reverse transcriptase inhibitor (NRTI), and non-NRTI (NNRTI) resistance mutations over time. In addition, we also detected recombinant forms and, for the first time, identified subtypes C, D, and CRF-24BG in PR. Recent studies suggest that non-subtypes B are associated with a high risk of treatment failure and disease progression. The constant monitoring of viral evolution and drug resistance mutation dynamics is important to establish appropriate efforts for controlling viral expansion.
Introduction
H
HIV-1 epidemics in the Caribbean region seem to be mediated by clade B. 23,24 Puerto Rico (PR), which is a commonwealth of the United States, has one of the highest prevalence rates of HIV-1 in the United States and the Caribbean. 25 In 2014, the prevalence of infection rate in PR was twice that of the average rate of the combined states and territories of the United States, ranking number 6 (567.3 per 100,000 population). 26 In the case of males, the most prevalent route of transmission is through heterosexual and male-to-male sexual contact. Among females, heterosexual contact is the most frequent mode of HIV transmission. 27 According to Los Alamos database, HIV-1 B is the most common variant and represents 99.7% of the virus subtypes in the infected population. 28 Recently, other subtypes and circulating recombinant forms (CRFs), including F(12-BF), A (01-BF), and CRF-39 BF-like, have been identified in the island. 29 Because of the high levels of migration to and immigration from the continental United States, it is important for the citizens of both areas to understand how the virus evolves. The purpose of this study was to assess the distribution of drug resistance mutations and subtypes in PR. Surveillance of HIV-1 genetic diversity is necessary to initiate public health efforts. 30
Materials and Methods
Ethics statement
The protocol was approved by the Institutional Review Board of Ponce Medical School Foundation.
Data source and variables
We analyzed 846 nucleotide sequences obtained from our secured “HIV Genotyping” test database, which contains de-identified sequence data from the years 2013–2017. The sequences were obtained from samples collected in PR between 2013–2017 and were amplified and analyzed as described in the subsequent sections. Descriptive statistics were used for demographic parameters.
Viral RNA extraction, reverse transcription polymerase chain reaction and sequencing
HIV-1 viral RNA was extracted using the QIAamp viral RNA kit (QIAGEN), following the manufacturer's instructions. Purified RNA was amplified by reverse transcription polymerase chain reaction (RT-PCR) using Titan OneStep RT-PCR kit (Roche). Briefly, first-round RT-PCR conditions were as follows: reverse transcription at 50°C for 10 min, inactivation at 94°C for 2 min, 35 cycles of 94°C for 10 s, 53°C for 30 s, and 68°C for 1 min; after the first 10 cycles, 5 s were added to the elongation step of each cycle, with a final extension at 68°C for 7 min. The first-round amplicon (2 μL) was reamplified using the Roche Fast Start PCR Mastermix. Second-round PCR conditions were as follows: 95°C for 15 min followed by 35 cycles of (94°C for 30 s, 53°C for 30 s, and 72°C for 2 min) and a final elongation at 72°C for 10 min. The sequences of the primers used for amplification and sequencing were as described in the World Health Organization (WHO) manual for HIV drug resistance testing using dried blood specimens. First-round primers: forward protease: 5′-TGAARGAITGYACTGARAGRCAGGCTAAT-3′; reverse protease: 5′-AYCTIATYCCTGGTGTYTCATTRTT-3′; forward RT: 5′-TTTYAGRGARCTYAATAARAGAACTCA-3′; reverse RT: 5′-CCTCITTYTTGCATAYTTYCCTGTT-3′. Second-round primers: forward protease: 5′-YTCAGRCAGRCCRGARCCAACAGC-3′; reverse protease: 5′-CTGGTGTYTCATTRTTKRTACTAGGT-3′; forward RT: 5′-TTYTGGGARGTYCARYTAGGRATACC-3′; reverse RT: 5′-GGYTCTTGRTAAATTTGRTATGTCCA-3′. Confirmed amplicons were directly sequenced using our WHO-accredited HIV genotyping protocols. Sequences were determined by using ABI 3730 XL. Sequences generated were uploaded onto our secured “HIV Genotyping” test database.
Analysis of drug resistance mutations and viral subtypes
Drug resistance mutations associated with protease inhibitors (PIs), nucleoside reverse transcriptase inhibitors (NRTIs), and non-NRTIs (NNRTIs) were determined by using Calibrated Population Resistance Tool (CPR) available in Stanford HIV Database program (
Phylogenetic analysis
The phylogenetic tree was inferred using the maximum likelihood (ML) method implemented by MEGA software v6. 34 We used bootstrap ML percentages to assess the robustness of each branch. Branches with bootstrap values above .70 were considered robustly supported. A reference set of 87 sequences was downloaded from REGA v3 database. The sequences were 1,030 bp long (nucleotides 2253–3452 relative to the reference sequence HXB2) and covered pol gene.
Results
During the period of 2013 through 2017, data were collected on 846 subjects. Demographic data show that the sample was predominantly male (74%) and the mean ages for males and females were 41 and 44 years, respectively (Table 1).
Age data available in 94% (males) and 89% (females) of the sequences, respectively.
CPR available from the Stanford HIV Database program was used to evaluate the percentage of expression of HIV-1 NRTI, NNRTI, and PI drug resistance mutations. 31 We detected a decrease in the prevalence of drug resistance mutations over time. In a period of 5 years, from 2013 to 2017, sequences with any PI, NRTI, and NNRTI SDRMs decreased 5%, 10.7%, and 6.6%, respectively (Fig. 1). However, each year, more than 21% of sequences had one or more drug resistance mutations (Fig. 1). Cross-class resistance, which involves two or three classes of resistance mutations, was observed in less than 10% of the samples. The drug resistance mutations associated with NRTI and NNRTI were detected more commonly than PI resistance mutations (Fig. 1). The most frequently observed PI drug resistance mutations during this period were L90 M and M46IL (∼14% each), whereas the most common NRTI and NNRTI drug resistance mutations were M184VI (30%) and K103NS (50%), respectively (Fig. 2).
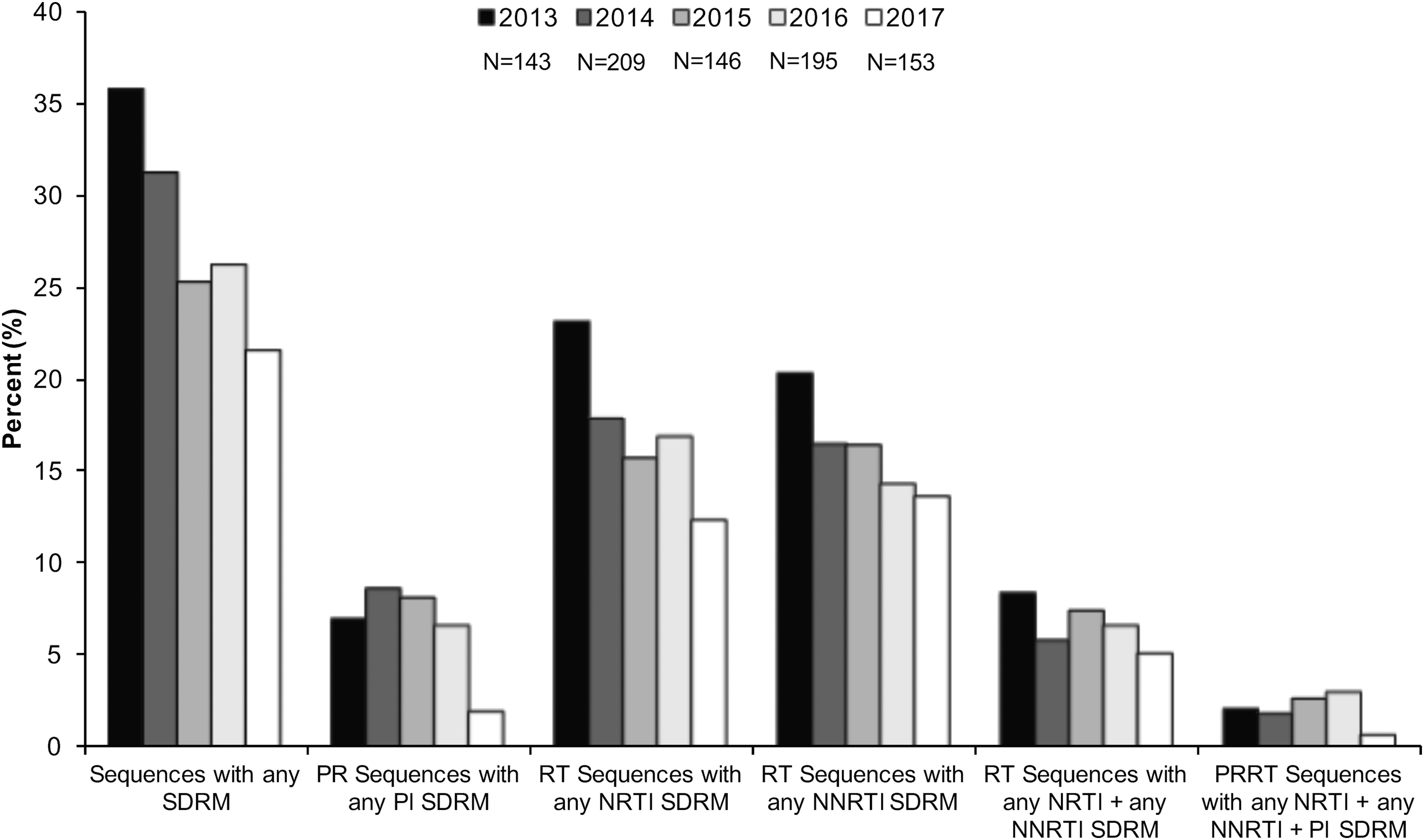
Prevalence of PI, NRTI, and NNRTI mutations over time. The prevalence of SDRM was evaluated using Calibrate Population Resistance Tool (CRP), Stanford HIV Database program. NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; SDRM, surveillance drug resistance mutations.
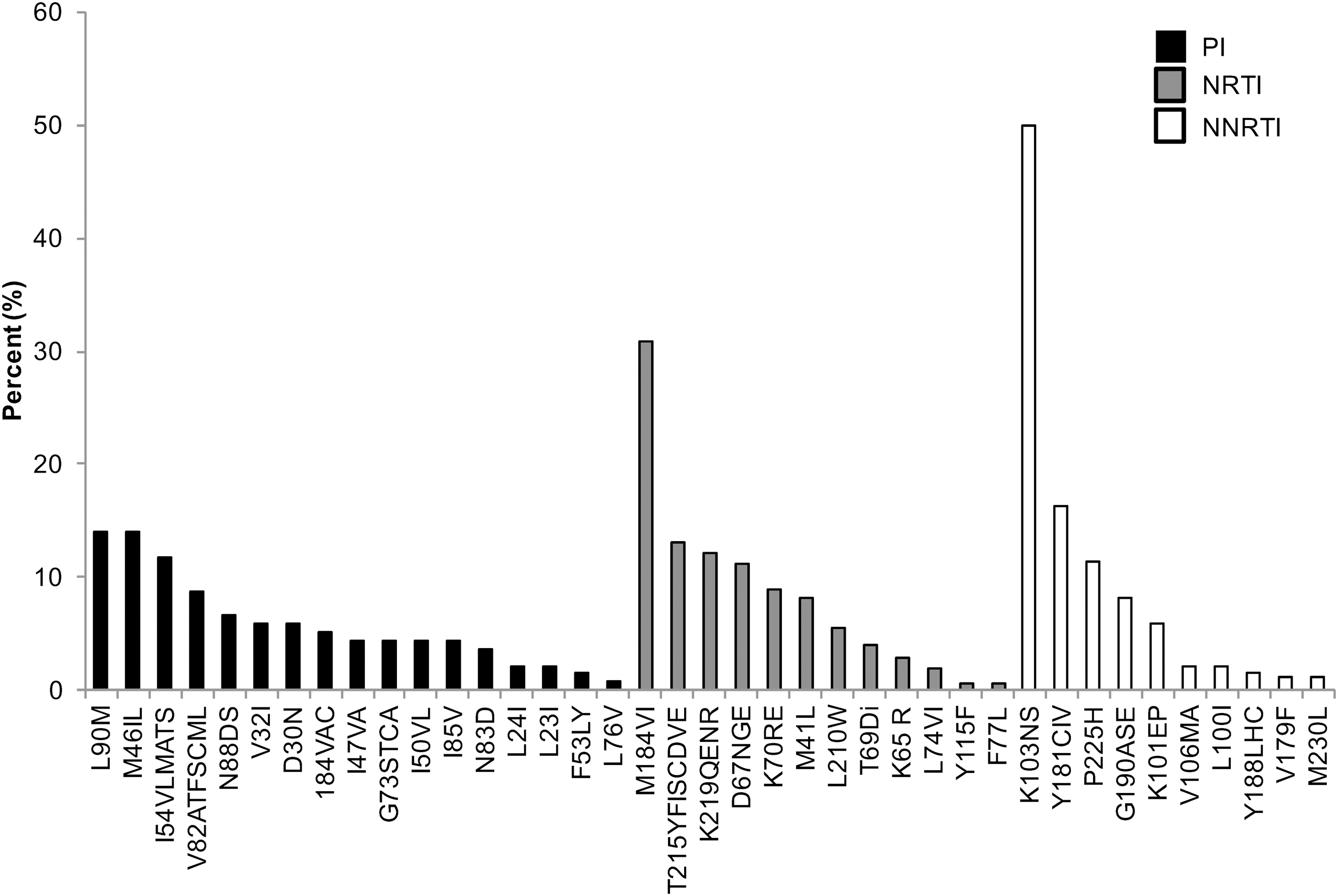
Frequency of specific HIV-1 drug resistance mutations among sequences analyzed during the period of 2013 through 2017. PI, NRTI, and NNRTI drug resistance mutations were evaluated using Calibrate Population Resistance Tool (CRP), Stanford HIV Database program.
Viral subtypes were assessed using the bootscanning method available in REGA v3 and confirmed using the COMET HIV-1 v2.2 and by jpHMM platforms. 32 The majority of HIV infections are associated with subtype B (n = 837; 98.9%). However, we detected other subtypes and recombinant forms in the island as follows: recombinant B/D (n = 1; 0.118%), F(12-BF) (n = 2; 0.236%), A(01-AE) (n = 2; 0.236%), A(A1) (n = 1; 0.118%), C (n = 1; 0.118%), D (n = 1; 0.118%), and CRF-24BG (n = 1; 0.118%) (Fig. 3). For the cases in which we obtained discordant results between platforms, we relied on the results obtained with the COMET platform for subtype assignment. Recent analyses suggest that COMET HIV-1 v2.2 has shown superior specificity in short sequences. 32,35 Despite the fact that subtype and inter-subtypes had been reported in PR [B/F, B/D, A1/B, A(A2), A(01-AE), F(12-BF), and CRF39-BF] previously, this is the first report of subtypes C, D, and CRF-24BG (Fig. 3). 29,36 The three cases were evaluated by using REGA v3, COMET HIV-1 v2.2, and by jpHMM available in jpHMM web server at GOBICS, to confirm the results 33,37 (Fig. 4). The breakpoint interval in CRF-24BG obtained by using jpHMM is located between the nucleotide positions 2498–2602 (based on HXB2 numbering). Subtype determination based on the pol sequences was visualized by using ML tree, generated using MEGA v .6, under a General Time-Reversible nucleotide substitution model (suggested by ModelTest) with a gamma-distributed rate variation for each of these tree alignments and a resampling process (100 bootstraps). 38,39 We compared samples processed in our site (n = 846) and reference sequences (n = 87, REGA v3 references samples). The three new cases reported in the island clustered with related sequences from around the world (Fig. 5).
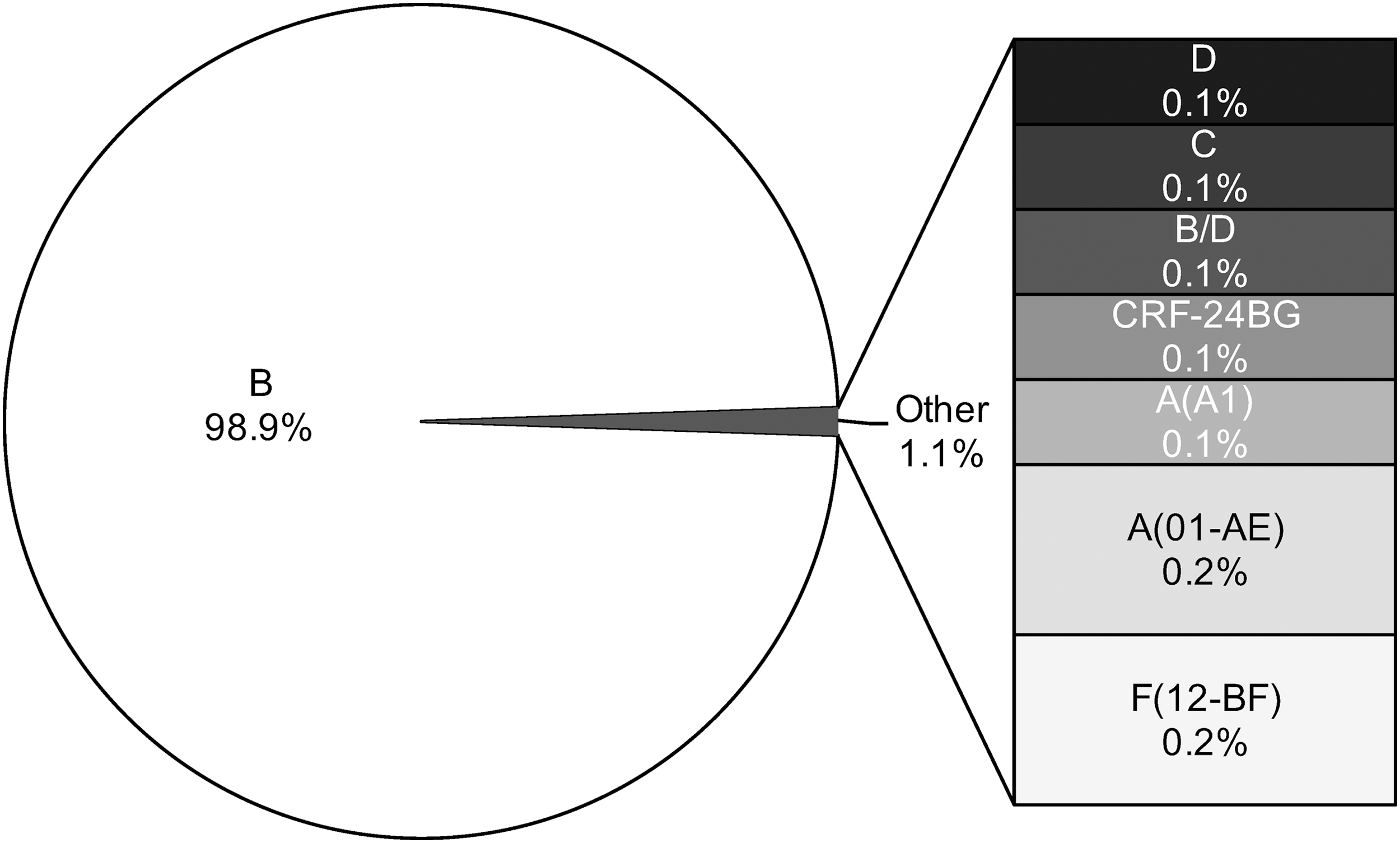
HIV-1 subtype assignment was performed using the bootscanning method within REGA v3 and confirmed using COMET HIV-1 v2.2 and jpHMM. Results indicate the percentage of a particular subtype represented in the sample (n = 846 sequences). COMET, Context-based Modeling for Expeditious Typing; jpHMM, jumping profile hidden Markov model.

Detection of novel HIV-1 subtypes in Puerto Rico.
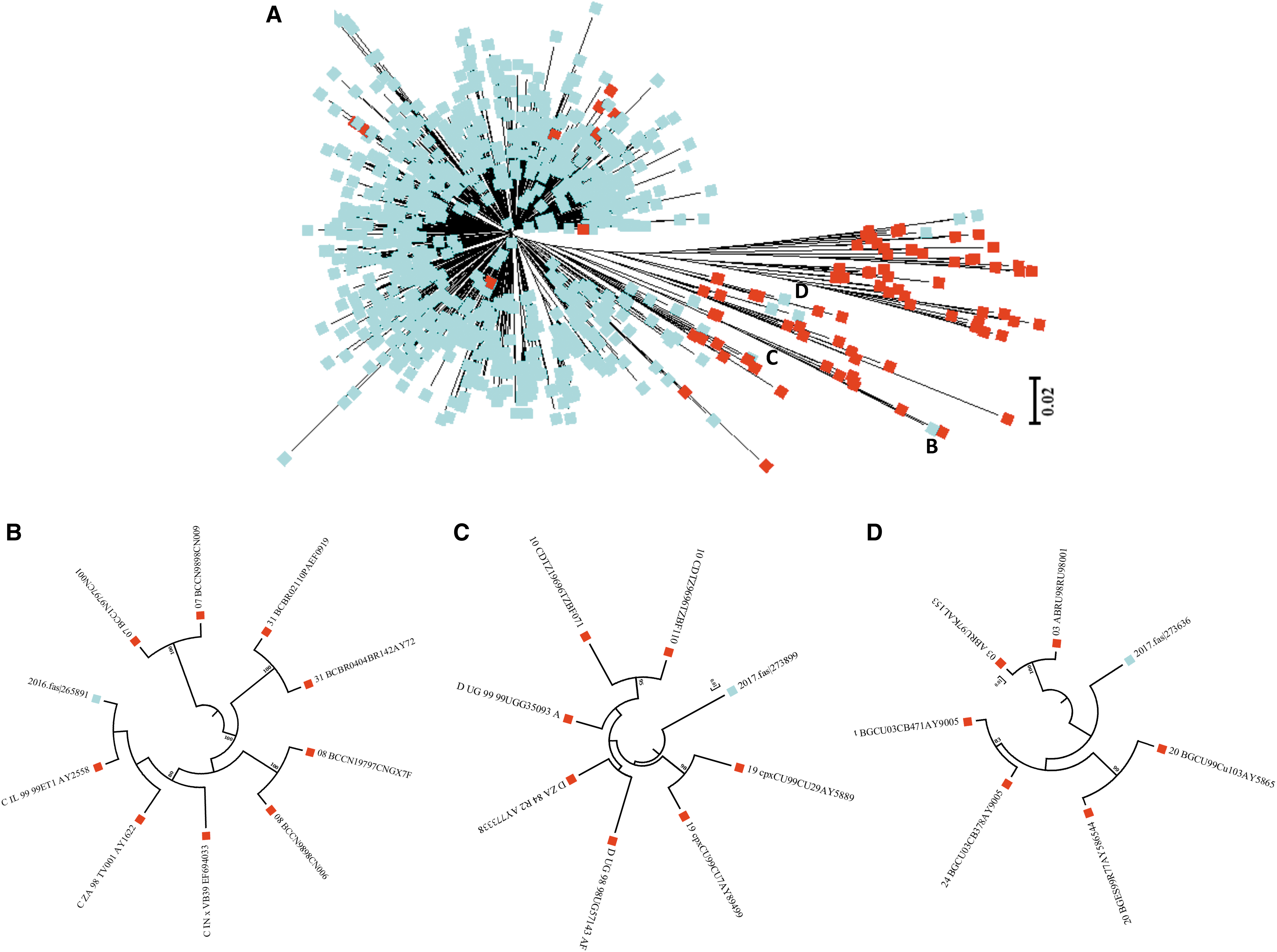
Maximum likelihood phylogenetic tree of HIV-1 pol gene (protease/reverse transcriptase) performed using MEGA software (v 6).
Discussion
After a positive diagnosis of HIV-1 infection, three different classes of drugs (NRTI, NNRTI, and PI) are used in different combination to treat patients. This treatment markedly inhibited replication and reduced transmission risk. 40,41 However, high viral turnover, error rate of the reverse transcriptase enzyme, poor adherence to medications, and selective pressure exerted by medications lead to genetic variants with drug resistance mutations. 42 The routine surveillance of the genetic diversity of HIV-1 virus is necessary to initiate public health efforts. The decline in PI, NRTI, and NNRTI mutations in PR (2013–2017) is consistent with recent studies that support that HIV-1 major drug resistance mutations are declining in resource-rich settings. 43,44 This finding would reflect the improvement of treatment regimens, leading to increases in treatment adherence. 44 –47 In addition, prevention programs that provide health insurance, care, and support to the HIV-1 infected patients have been established effectively by local health departments and nongovernmental organizations.
Our finding that M184VI and K103NS were the most frequent reverse transcriptase mutations is in agreement with results obtained by Sepulveda-Torres et al., who performed an analysis on PR samples during the period of 2002–2011 with data generated using TRUGENE HIV-1 Genotyping Kit and OpenGene DNA sequencing system. 48 In their study, M184VI and K103NS were also identified as the predominant drug resistance mutations. The M184VI resistance mutation is an NRTI-resistance mutation associated with high level of resistance to lamivudine and emtricitabine and cause low level of resistance to abacavir and didanosine. 49 –51 However, treatment with lamivudine and emtricitabine increases susceptibility to zidovudine, tenofovir, and stavudine and is associated with reduced HIV-1 replication. 52 –58 The K103N is an NNRTI resistance mutation associated with reduced susceptibility to nevirapine and efavirenz; however, K103S causes high-level resistance to nevirapine and intermediate resistance to efavirenz. 57,59 –62 In addition, K103N shows a viral fitness, which is the ability of the virus to adapt and reproduce in the host, very close to levels comparable to that of the wild-type virus. 63 The highest PI mutations were L90M and M46IL, which are associated with significant reductions in fitness. 63 –66 The L90M is associated with reduction of susceptibility of PI medications, except tipranavir and darunavir. 67,68 The M46IL mutations occur alone or in combination with others, which are associated with reduced susceptibility to atazanavir, fosamprenavir, indinavir, lopinavir, and nelfinavir. 69,70 The high prevalence of L90M and M46IL mutations in our study is similar with the results obtained in the INSIGHT Strategic Timing of ART trial. 71 The finding that reverse transcriptase drug resistance mutations were more commonly expressed than resistance mutations to PI has been observed in other studies. The increase in reverse transcriptase drug resistance mutations may be related to the elevated use of NNRTs/NNRTIs, and in the case of NNRTI resistance mutations, to the increased transmission in newly diagnosed patients, or to the interruption of a suppressive NNRTI-based regimen. 44,71 –73 A limitation in this study is unavailability of sequence data to assess integrase strand inhibitor resistance mutations for all sequences under the study period (2013–2017). Not all sequences analyzed in this study included integrase gene sequences. Thus, additional studies are needed to evaluate the molecular evolution of HIV-1 integrase at the interpatient level to assess the ongoing adaptation of the enzyme.
PR has one of the highest prevalence rates of HIV-1 in United States, with male-to-male and heterosexual transmissions as the major routes of the infection. While the HIV-1 epidemic affecting the Caribbean is largely mediated by B-clade virus, 74 –76 only a small number of non-B-clade HIV-1 infections have thus far been identified in the region. 29,36,76 Although a previous study demonstrated that HIV-1 isolates from PR were closely associated with U.S. B-clade HIV-1 strains, the Caribbean B-clade HIV isolates are distinct from the U.S. B-clade isolates. 23,77 Cuba is an exception, with high genetic diversity and circulation of several viral variants of non B-clade. 78 To gain information that may be useful for developing strategies to prevent further spreading of HIV-1 epidemic in PR, it is important to focus our efforts on identifying the viruses circulating through the island. Understanding of how the virus evolves is crucial due to high levels of migration to and immigration from the continental United States. In the Caribbean region, subtype C has been previously reported in Cuba and Saint Lucia, while subtype D and CRF-24BG have been reported only in Cuba. 28,76,79,80 Our current report of the first cases of subtype C, D, and CRF-24BG in PR highlights the importance of molecular monitoring of new subtypes spreading in the island.
The most relevant biological properties among each HIV subtypes are their rate of adaptive evolution, neutral mutations, tropism, and acquisition of antiretroviral resistance. 6,81 –83 In addition, transmissibility of the virus and patient's response to antiretroviral treatment have been implicated with the HIV-1 genetic subtype classification. Subtype C, which is the most prevalent subtype in the world (nearly 50%), is very common in Southern Africa and India. 7 This subtype has been associated with increased infectivity during heterosexual intercourse and poorer ART outcomes, and develops resistance to antiretroviral therapy faster than other subtypes. 84 –87 Recent studies suggest that subtype D may be associated with a faster virological rebound, CD4 decline, prevalence of CXCR4-using virus, and high rates of disease progressions. 88 –91 The CRFs are increasing in the past decade in various parts of the world. 92,93 The CRF-24BG was previously reported in Cuba and other recombinant BG strains in Portugal, Spain, and Germany, which were associated with intravenous drug users. 94 –96
The possible role of tourism in the spread of non B-clade strains needs to be addressed. Recent studies have emphasized the important role of tourism in spreading the HIV-1 epidemic to and throughout the Caribbean. 48,97 –99 In addition, the migration of the HIV-1 at-risk population among the different countries is undoubtedly related with the flow of the epidemic. 100 –102 The establishment of local and regional (Caribbean) programs to monitor how the virus evolves in our countries can help us for developing strategies to prevent the HIV-1 epidemic from spreading in the Caribbean region. An example of unusual mutations in the Caribbean region is the signature of threonine deletion (env gene) in Trinidad and Tobago, which is predominantly associated with heterosexual transmission. 103 The circulation of HIV-1 non-B clade in the island, which favors recombination, may have repercussions in diagnostic and clinical management of HIV-infected individuals. 104 –106 While epidemiological and clinical information about patients infected with these novel subtypes were not available for this study, our current analysis provides new data that will be useful for understanding how the HIV-1 virus evolves in PR. This information can help us develop strategies to prevent the impact of HIV-1 epidemic spread in PR into a more complex epidemiological landscape.
Footnotes
Acknowledgments
The study was supported by the AIDS Research Infrastructure Program (RCMI Program; NIMHD-G12-MD007579).
Results included in this article were presented at the Research Center in Minority Institutions Translational Science Conference, October 28–November 1, 2017.
Sequence Data
The sequences are available at GenBank with accession numbers MF960930-MF961775.
Author Disclosure Statement
No competing financial interests exist.