
Abstract
HIV-1 group N (HIV-1/N) remains rare and mainly restricted to Cameroon. In this study, we report a new HIV-1/N infected case identified during routine HIV screening activities in Yaounde. The genetic characterization of the near full-length genome of this virus strain revealed that it is genetically distinct to all HIV-1/N described to date. However, the Vpu protein responsible for tetherin antagonism displayed the same amino acid substitutions (E15A, V19A, I25L, and V26L) as other HIV-1/N from Cameroon.
H
In this study, we report a new HIV-1/N infected case identified during routine HIV screening activities in Yaounde. In the Virology Service of Centre Pasteur of Cameroon, HIV type/group is routinely determined using an in-house ELISA assay based on peptides derived from the immunodominant region of env-gp41 and the V3 loop of env-gp120 as previously described. 5 Among the 2,416 HIV-positive samples identified in 2014 in our laboratory, 1,931 (80%) were classified using serotyping as HIV-1/M, 12 (0.5%) as HIV-1/O, 10 (0.41%) as HIV-1/M+O, 2 (0.08%) as HIV-2, and 1 (0.04%) as HIV-1/N. About 460 (19.04%) samples did not react with any peptide.
Due to cross-reactivity that could be observed between HIV-1 groups M and N, molecular tests were performed to confirm HIV-1/N reactivity obtained with the serotyping assay. Therefore, reverse transcriptase-polymerase chain reactions were performed in the gp41 and Pol regions, and phylogenetic analyses of the sequences generated from the two genomic regions indicated that the patient was infected with an HIV-1/N. The characterization of the near full-length genome sequence (9,104 bp; accession No.: MF767262) determined by sequencing and assembling of overlapping fragments as previously described 6 confirmed a phylogenetic link to HIV-1/N all along the genome. Interestingly, it was more closely related to the strain YBF106 in the phylogenetic tree suggesting that the two strains could be genetically similar (Fig. 1b, c). However, the similarity plot analysis of the near full length showed that the similarity with YBF106 was higher in the Gal-Pol region but lower in the Env region (data not shown), probably due to the immune pressure that led to the great diversity overserved in the HIV-1 envelope region compared to other genomic regions. YBF106 was described in 2004 in a 56-year-old man who died shortly after sample collection, while the newly characterized HIV-1 strain was obtained from a 43-year-old Cameroonian woman living in the center region of Cameroon whose husband died just 6 years ago before she was received at Centre Pasteur of Cameroon for HIV screening in 2014. Therefore it is very difficult to find an epidemiological link between both individuals to check for a transmission pair.
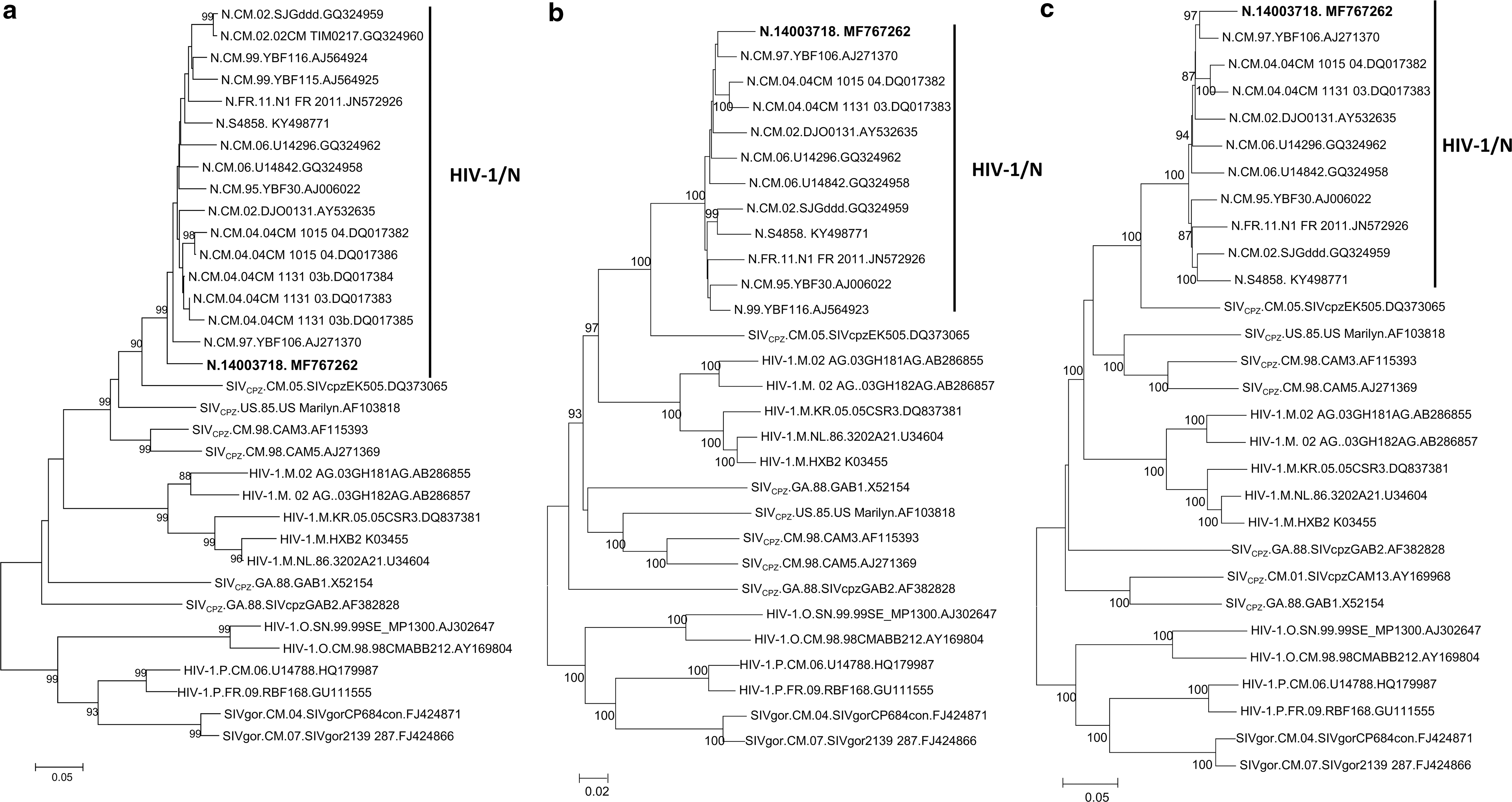
Phylogenetic trees of gp41, Integrase, near full-length genome, and Vpu amino acid sequences of the new HIV-1/N strain. Phylogenetic trees of (

The new patient was naive for combined antiretroviral therapy (cART) and her CD4 cell counts were 276 cells/μL. A high plasma viral load (5.6 and 6.1 log copies/mL) was obtained with the Abbott Real-Time HIV-1 assay (Abbott Molecular, Wiesbaden, Germany) and the Generic HIV viral load assay (Biocentric, Bandol, France), respectively, indicating that the virus was highly replicant competent. The resistance profile of the newly sequenced HIV-1/N was investigated based on the pol sequence (Prot-RT) and using the French ANRS resistance algorithm (
It was previously reported that HIV-1/N may have a weaker capacity to antagonize the human restriction factor tetherin than pandemic HIV-1/M strains, what could be responsible of the epidemiological differences between HIV-1/M and HIV-1/N. 9 To assess the ability of this new HIV-1/N to antagonize the tetherin, we compared its Vpu protein sequence to those of other HIV-1/N strains characterized to date, as well as the Vpu protein sequence of the SIVcpz strain EK505 which is one of the HIV-1/N precursors. 10 The results showed that the Vpu protein sequence of the new HIV-1/N described in this study was closer to those N-Vpu proteins from Cameroon than the strain N1FR2011 from Togo (Fig. 1d); this latter having an anti-tetherin profile similar to that of HIV-1/M strains. 9 The four amino acid substitutions (E15A, V19A, I25L, and V26L) described by Sauter et al. that could allow efficient interaction with human tetherin in HIV-1/N were also present in the new strain. 9 However, they found that despite these changes, HIV-1/N strains from Cameroon still have a weaker capacity to antagonize the human restriction factor tetherin 9 probably because they exhibited aberrant intracellular localization and failed to recruit the enzymatic complex that induces tetherin degradation.
The anti-tetherin profile of the new strain contrasts with the high plasma viral loads found with two different assays. This high level of in vivo replication confirms previous studies showing that HIV-1/N in vivo and in vitro replicative capacity could be similar to HIV-1/M despite difference of Vpu efficiency. 8,11 Therefore, lack of diffusion is probably multifactorial rather than only due to anti-tetherin property. HIV-1 groups M and N have been suggested to have derived from the two distinct SIVcpzPtt viral lineages and geographically isolated chimpanzee communities: south eastern Cameroon for HIV-1/M and south central Cameroon for HIV-1/N. However, these two HIV-1 variants have different epidemiology as follows: HIV-1/M is found worldwide, while HIV-1/N has remained rare and restricted to Cameroon. Accordingly, the rate of HIV-1/N, found to be at 0.04% in this study, suggests that the prevalence of HIV-1/N continues to be very low in Cameroon. However, it should be noted that this group is one of the two most recent (1963) HIV-1 groups in the human population compared to HIV-1 groups M (1908) and O (1930). 12 Thus, there is no information if HIV-1/N is still in the phase of adaptation to the human population or not.
Discrimination between HIV-1 groups is mainly based on serotyping which is performed only in some reference laboratories in Cameroon. The HIV-1/N described here elicited a strong reactivity with HIV-1/N peptides, and this result was confirmed by molecular analysis of different subgenomic regions, as well as the near full-length genome sequence. This suggests that serotyping could be an effective tool to identify HIV-1/N infections, despite the serological cross-reactivity that can occur between HIV-1 group M and N antibodies.
In conclusion, our results suggest that HIV/N continues to circulate in Cameroon even though its prevalence remains consistently low. Therefore, HIV types/group determination using serotyping should be performed on all HIV-positive samples to document the potential trends in the relative proportions of HIV-1/N and other HIV variants circulating in Cameroon.
Patient Consent Form
Written informed consent was obtained from the patient, and a copy is available if required.
Footnotes
Acknowledgments
The authors thank Centre Pasteur of Cameroon and Rouen University Hospital for the financial support for the molecular characterization of the new virus strain. The authors are also grateful to the patient who consented to participate in this study.
Authors' Contributions
J.C.P., R.N., and A.K. conceived and designed the study. P.A.N.T., S.A.S.M., F.D.O., and E.N.M. carried out experiments. P.A.N.T., S.A.S.M., and E.N.M. drafted the article. F.D.O, L.N., J.C.P, R.N., and A.K performed data analysis and reviewed the article. All the author
Sequence Data
GenBank accession number for the near full-length genome sequence reported in this study is MF767262.
Author Disclosure Statement
No competing financial interests exist.