
Abstract
Some HIV-associated complications involve mitochondrial dysfunction and may be less common in individuals with iron-loading HFE (hemochromatosis gene) variants. We evaluated HFE 845A and 187G alleles in relation to mitochondrial DNA (mtDNA) levels in peripheral blood mononuclear cells from 85 individuals with HIV infection on uninterrupted antiretroviral therapy (ART) for 15 or more consecutive weeks. Carriers of HFE gene variants (N = 24) had significantly higher mtDNA levels than noncarriers (N = 61), after adjusting for age, race, sex, and type of ART [adjusted β-coefficient 297, p-value < .001 for at least one HFE variant], but mtDNA declined among all individuals on study during 48 weeks on ART. Increased cellular mtDNA content may represent a compensatory response to mitochondrial stress that is influenced by iron-loading HFE variants.
Introduction
M
In non-human primates, abnormal mitochondrial morphology and respiratory enzyme activity have been linked to nucleoside reverse-transcriptase inhibitor (NRTI)-associated mtDNA depletion. 5 Accumulating evidence now indicates that the mitochondrial pathogenesis of many long-term complications of HIV infection in fact derives from multiple factors, not only from the inhibition of mtDNA pol-γ. These factors include HIV infection itself, direct cytokine-mediated effects on mitochondria, and other antiretroviral drug effects on these organelles. 6
Depletion of mtDNA, frequently studied in peripheral blood mononuclear cells (PBMCs), has been reported in HIV-seropositive (HIV+) individuals who are treated with NRTIs, particularly those exposed to dideoxy-NRTIs such as didanosine (ddI) or stavudine (d4T). Moreover, in ART-naive individuals with HIV, mtDNA depletion has been observed in both B and T cells; it correlates negatively with expression of immune-activation markers, and mtDNA levels in B cells correlate with the rate of CD4+ T-lymphocyte loss. 7,8 The hierarchy of clinical NRTI toxicity parallels the degree of inhibition of DNA pol-γ by these drugs. 9 Early studies documenting the loss of mtDNA in PBMCs from treatment-naive individuals with HIV also suggested that direct effects of HIV on mitochondrial function and/or oxidative stress may be important and that these effects are further compounded by use of older, more mitochondria-toxic NRTIs. 10 While the older and most toxic NRTIs are seldom used today, all NRTIs have some affinity for mtDNA pol-γ, and contemporary NRTIs also have measurable effects on mitochondrial function in clinical studies; even other classes of ART can have mitochondrial effects. 11 –13 Hence, it remains important to advance understanding of the basis of HIV- and ART-related mitochondrial dysfunction.
Iron is an essential micronutrient and cofactor for a large number of metabolic enzymes, and a steady supply of cellular iron is needed for normal mitochondrial function and cellular energy production. Accumulating evidence also suggests that mitochondrial function and mtDNA variants play a critical role in cellular iron homeostasis and inflammation. 14 –17 The HLA class I-like protein encoded by the hemochromatosis (HFE) gene affects macrophage function and inflammatory responses, partly by influencing hepatic synthesis of the master iron-regulatory, proinflammatory hormone, hepcidin. Hepcidin modulates systemic iron absorption and monocyte-macrophage iron content in response to a variety of inflammatory signals. Monocyte-macrophage iron content is important in oxidative microbial killing and may significantly influence proinflammatory cytokine production by cells of this lineage. 18,19
We previously reported associations of common variants in HFE, G845A and C187G, with a significantly reduced incidence of both peripheral sensory neuropathy and lipoatrophy during ART in AIDS Clinical Trials Group (ACTG) Study 384 participants. 20,21 The mechanisms for those associations potentially include modulating effects of HFE-mediated increases in systemic iron levels on ART-induced mitochondrial toxicity. The objective of this study was to evaluate the impact of HFE variants on mtDNA content in PBMCs among persons who were receiving chronic, uninterrupted ART that included antiretrovirals with known mitochondrial toxicity.
Methods
Study design and subject selection
DNA samples and linked clinical data for this nested case-control analysis were collected from 87 participants in ACTG protocol A5117 (NCT00017771), in whom mtDNA copy number per cell measurements was available and who also provided DNA for genetic studies. ACTG Study A5117 was a prospective, multicenter, observational cohort study of peripheral neuropathy in HIV infection, which enrolled individuals with HIV infection between October 2001 and June 2002 at participating sites of the ACTG and Neurologic AIDS Research Consortium (NARC) with neurological expertise. 22 All subjects provided written informed consent, and the institutional review boards of all participating centers approved the study. Eligible subjects were 13 years of age and older with advanced HIV disease, as evidenced by a CD4+ T-lymphocyte count <300 cells/mm3, and at least 15 consecutive weeks of ART exposure. Of note, individuals with conditions other than HIV infection or a history of ART that might confound the diagnosis of neuropathy related to ART were excluded. Detailed inclusion/exclusion criteria and study procedures for ACTG study A5117 were previously published. 22
Genotyping and mtDNA measurements
Whole-blood samples were collected at study entry and at 48 weeks on study in a 10-mL acid citrate dextrose tube (Becton Dickinson, San Jose, CA), and lymphocytes were isolated by Ficoll gradient centrifugation. Total genomic DNA was extracted from blood-derived lymphocytes [QIAamp DNA Blood Mini kit and DNeasy Tissue kit, both from Qiagen, Inc., Valencia, CA]. HFE genotypes at the two most commonly polymorphic loci, 845G>A, or G845A (rs1800562) and 187C>G, or C187G (rs1799945), were determined using the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems) and customized TaqMan assays, as reported elsewhere. 20
Dry pellets of lymphocytes (1 × 107 cells) were stored at −70°C. Total DNA was assayed for mtDNA using mitochondrial primers that amplify a 90-bp region of mtDNA encoding NADH dehydrogenase subunit 2, and genomic DNA primers specific for the region encoding Fas ligand (98 bp), by real-time PCR, using published PCR cycling conditions. The PCR reactions were assayed with the Lightcycler FastStart DNA Master SYBR Green I mix in the LightCycler real-time instrument (Roche Diagnostic Corporation, Indianapolis, IN). Each sample and standard were run in duplicate (20-μL reaction volume containing 1× SYBR Green master mix, 0.5 μM of each primer, and 10 ng of nuclear [genomic] DNA [nDNA]). Results were analyzed with LightCycler Version 4.0 software. The number of mtDNA copies per cell was calculated using the following formula: (mtDNA copies/nDNA copies) × 2 = mtDNA copies per cell. 23 All mtDNA/nDNA measurements for this analysis were performed by a single laboratory, eliminating the intersite assay variability that has previously been reported. 24
Statistical analyses
The distribution of variables at entry onto the study was compared between HFE genotype categories by the Student's t-test or Mann–Whitney U test (continuous variables), or by the chi-square or Fisher's exact test (categorical variables). Unadjusted analyses of mtDNA copies per cell at entry and at 48 weeks of observation were performed using the nonparametric Mann–Whitney U test, because this variable was not normally distributed, and results were stratified by self-reported race/ethnicity and HFE genotype. Multivariable analyses of the association between HFE genotype and mtDNA copies per cell at entry, mtDNA copies per cell at 48 weeks, and the change in mtDNA copies per cell over 48 weeks on study were performed by multiple linear regression, adjusting for age, race/ethnicity, sex, ddI use, total dideoxy-NRTI drug (d-drug) use [0 vs. ≥1 d-drug at entry], CD4+ T-lymphocyte count, and HIV RNA concentration at study entry. STATA statistical software version 10.0 was used for all analyses (Stata Corp LP, College Station, TX).
Results
Baseline demographic and clinical characteristics of study subjects are shown in Table 1. The mean age of HIV+ persons was 37 ± 9 years, 8 (9%) were women, and 39% were non-white. This population had advanced HIV disease, with a mean CD4+ T-cell count of 165 per mm3 and a median HIV RNA concentration in plasma (viral load) of 2.9 log10 (copies per mL). Treatment regimens at study entry included ddI in 22%, d4T in 33%, and zalcitabine (ddC) in 1.1% of subjects, and median cumulative ddI exposure was 1.4 years (range 0–6.6 years). Use of at least one NRTI drug was reported at the baseline visit in 82 (94%) and protease inhibitor use in 60 individuals (69%); prevalence of non-NRTI (NNRTI) use was 37%. Median cumulative exposure to other d-drugs in the study sample was 2.1 years for d4T (63 individuals, range 0–7.2 years) and 10 months for ddC (15 individuals, range 2 months to 5.2 years); however, data on the duration of all prior ART among participants on ART at entry were not available.
Two individuals could not be genotyped at the HFE G845A or C187G loci.
Only self-reported race differed significantly by HFE genotype (p < .01).
Information on cumulative d-drug use was available in only 45% of individuals; p-value for comparison of ancestral versus variant HFE allele categories was not statistically significant.
HFE, hemochromatosis 845A and/or 187G variant allele(s); SD, standard deviation; ddI, didanosine; d4T, stavudine; ddC, dideoxycytosine; NRTI, nucleoside reverse-transcriptase inhibitor; NNRTI, non-nucleoside reverse-transcriptase inhibitor; PI, protease inhibitor.
Among 87 study participants with available DNA and mtDNA copies per cell measurements, HFE G845A and C187G genotypes were successfully determined in 85 (98%); there were no G845A or C187G homozygotes, eight 845GA heterozygotes, 16 C187CG heterozygotes, and no compound heterozygotes. Occasional or frequent alcohol use was reported by 65% of participants and was similar in both genotype groups. Carriers of one variant HFE allele (187G or 845A) were not significantly different from HIV+ individuals with neither HFE variant (noncarriers) with respect to age, sex, and other baseline characteristics (including individual d-drug and cumulative ddI use), except for race/ethnicity. As anticipated from the known racial distribution of the HFE variants studied, individuals with HFE variant alleles were significantly more likely than noncarriers to be white (86% vs. 49%, p < .01).
Results of unadjusted analyses of mtDNA copies per cell at baseline in subjects with and without HFE variants are shown in Table 2. Median mtDNA content at entry in PBMCs was significantly higher among all 24 subjects with at least one HFE variant than among the 61 individuals with ancestral alleles at both HFE loci [668 (interquartile range, IQR 324, 954) vs. 396 (IQR 274, 537) copies per cell, respectively; p = .01]. Similarly, significant differences were observed in whites [median mtDNA copies per cell of 613 (IQR 335, 897) copies per cell among 21 carriers of HFE variant alleles vs. 344 copies per cell (IQR 231, 447) among 30 noncarriers, p = .03]. Median mtDNA copy number per cell was significantly higher in carriers than in noncarriers of HFE variant alleles among whites (p = .03). The median absolute change in within-individual mtDNA copies per cell from study entry (baseline) to 48 weeks on ART was also greater for subjects with HFE gene variants in the entire study sample, although this difference did not reach statistical significance [−314 (IQR −662, −3) in individuals with at least one variant vs. −108 (IQR −187, 53) in individuals with only ancestral HFE alleles, corresponding to −47% vs. −19% percent change, respectively (p = .08)].
p-values <.05 are considered statistically significant.
Longitudinal mtDNA copy number data were available in 86% of participants.
HFE, hemochromatosis gene; mtDNA, mitochondrial DNA; IQR, interquartile range; BL, baseline.
Figure 1 summarizes mtDNA copy numbers at entry and 48 weeks, stratified by HFE genotype status. The decline in PBMC mtDNA content was also nonsignificantly greater among whites with HFE variants compared to those without HFE variants [median −314 (IQR −662, 3) vs. −116 (−211, 70), p = .15], corresponding to a −47% and −25% change, respectively. MtDNA copy number per cell was nonsignificantly higher at baseline in participants with HFE 845A or 187G alleles, when these alleles were evaluated separately, but the number of individuals with HFE 845A alleles was very small; differences between 187G carriers and noncarriers approached statistical significance (p = .07, Table 3).
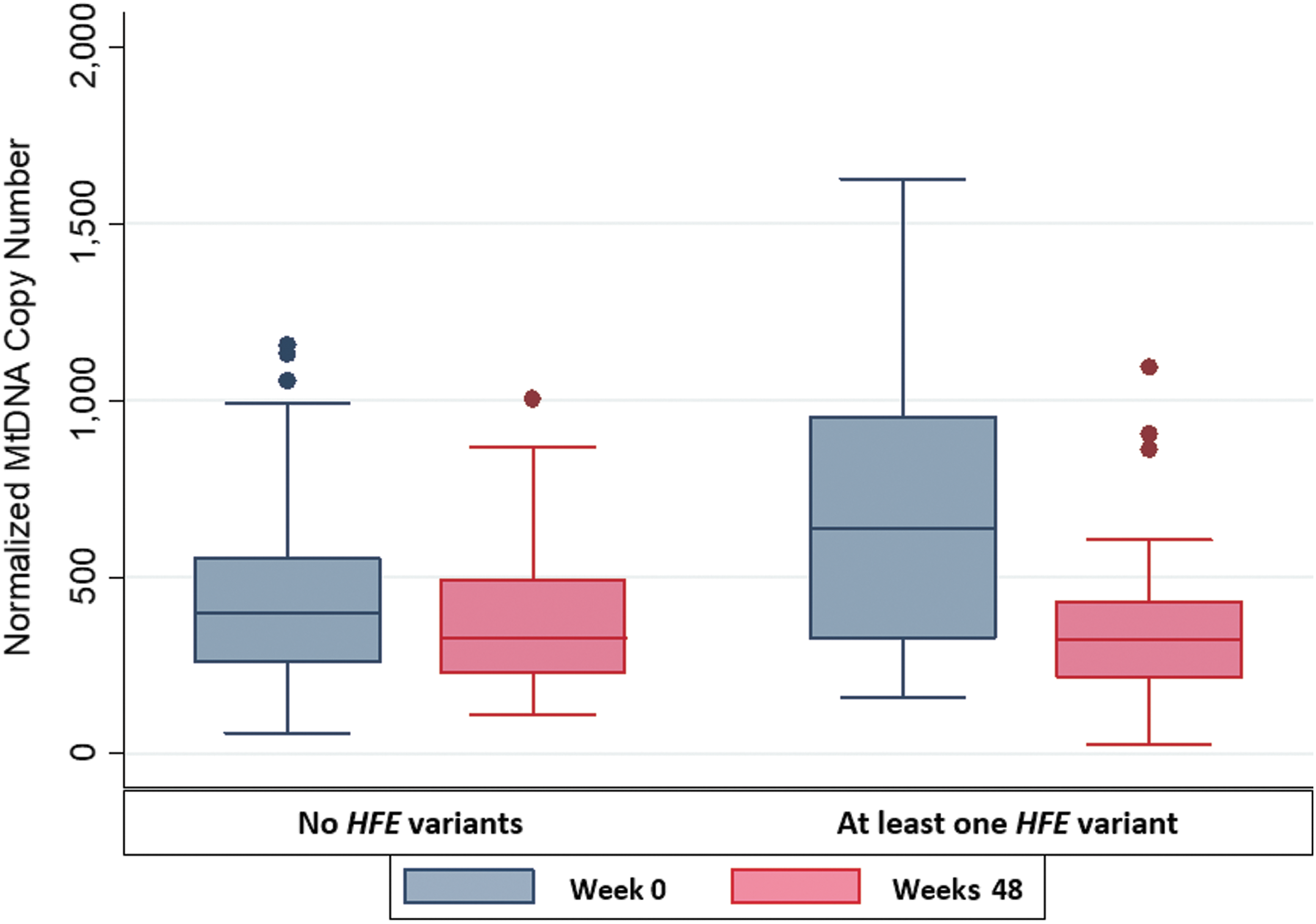
Mitochondrial DNA copy number per cell at entry and 48 weeks, stratified by HFE genotype status. HFE, hemochromatosis gene.
Unadjusted p-values for comparison of mtDNA copy number by HFE G845A and C187G genotype are shown. p-values<.05 are considered statistically significant.
Protease inhibitor use, mitochondrial haplogroup T status (previously linked to NRTI-associated mitochondrial toxicities), and d4T and ddC use at baseline were not significantly associated with either mtDNA copies per cell or with the presence of HFE variants (some of these data not shown), and were therefore not included individually as covariates in multivariable regression models; however, ddI use and the total number of NRTIs used were included. MtDNA copies per cell in study participants at baseline were not associated with CD4+ T-cell count or HIV RNA concentration in plasma.
Multivariable regression analyses revealed that HFE variant alleles were independently associated with greater PBMC mtDNA content at baseline (β-coefficient 297, p < .001) in this extensively ART-treated population, adjusting for age, self-reported race/ethnicity, sex, and the following variables: total NRTI drug use, ddI use, CD4+ T-cell count, and plasma HIV RNA concentration (results presented in Table 4). Associations of mtDNA copy number with ddI use and with total NRTI use tended toward statistical significance [β-coefficients −165 (p = .05) and 67 (p = .08), respectively], whereas no associations were observed with CD4+ T-cell count or plasma viral load (β-coefficients −0.42 (p = .26) and −36 (p = .20), respectively].
For each factor in the model, the estimate of association with mtDNA copy number [β-coefficient, 95% confidence interval (95% CI)] is shown; p-values <.05 are statistically significant.
A stratified analysis among 51 self-described white participants was also performed, since HFE variants are considerably more prevalent in whites, and the results of this analysis were similar. Median mtDNA copy number per cell at baseline was 613 (IQR 335, 897) among white individuals with HFE variants (845A or 187G), compared to 344 (IQR 231, 447) in noncarriers of these variants (p < .05).
Discussion
This study evaluated potential associations between PBMC mtDNA content and common HFE G845A and C187G missense variants. Importantly, susceptibility to the mitochondrial toxicities of ART is still relevant in the modern ART era: antiretroviral drugs with mitochondrial effects continue to be used in low-resource settings due to their lower cost, and the possibility of subclinical mitochondrial toxicity during treatment with antiretrovirals other than the dideoxy-NRTIs remains an open question. 25,26 Previous studies have documented both increases and decreases in mtDNA levels as a physiological response to mitochondrial stress 27 –31 and HFE variants have been associated with elevated urinary levels of F2-isoprostanes (a specific marker of in vivo oxidative injury), 32 as well as with indicators of increased mitochondrial stress, 33,34 such as reduced mitochondrial membrane potential. This study represents the first report of an independent association of HFE variant genotype with increased mtDNA copies per cell in PBMCs from individuals with HIV on ART.
The NRTI drug class is known to inhibit mtDNA polymerase-γ, and mtDNA depletion could play an important role in mediating or promoting mitochondrial toxicities in clinical practice. 6,35 The finding that mtDNA copies per cell during ART decreased over the course of follow-up for all participants, regardless of HFE genotype, provides confirmatory evidence of ART-mediated mitochondrial stress. Responses to this stress may, in turn, be influenced by changes in iron transport or other downstream effects of HFE variants, such as altered macrophage-mediated inflammation. 36 The impact of HFE variants on the risk of neuropathy and other mitochondrial sequelae of ART may therefore be influenced by a combination of HFE-mediated changes in systemic iron levels, cellular iron transport, and cellular mtDNA content.
Prior work has shown that individuals heterozygous at the HFE locus have increased systemic levels of iron, as well as oxidatively active (non-transferrin bound and labile) forms of iron, and they experience higher levels of oxidative stress. 37 –40 Hence, it would not have been surprising to find that HFE variants increased the risk of mitochondrial toxicities in individuals with HIV. To the contrary, we previously reported the association between HFE variant G845A (Cys282Tyr) and a decreased risk of NRTI-associated peripheral neuropathy in ACTG study 384. 21 The more prevalent polymorphism C187G (His63Asp) is also associated with higher systemic iron indices and was associated with reduced risk of neuropathy among whites in univariate, but not in multivariable-adjusted analyses. The 187G SNP was also significantly associated with a reduced risk of lipoatrophy, another ART-associated mitochondrial toxicity. 20 A possible explanation for these associations is that increased mtDNA synthesis occurs as a physiological response to mitochondrial stress or toxicity during ART in individuals with higher iron stores, as iron is essential for normal mitochondrial function and energy metabolism, and iron metabolism and mitochondrial metabolism are coordinately regulated. 41 –44
Considerable evidence supports the depletion of mtDNA content in HIV- and ART-related comorbidities, such as lipoatrophy, diabetes mellitus and other metabolic disorders, increased risk of cardiovascular and hepatic disease, and accelerated aging. 25,45 –47 In addition, it has also been reported that pregnant women with HIV have significantly lower mtDNA/nDNA ratios than pregnant women without HIV infection after controlling for platelet count, raising the possibility that lower levels of maternal mtDNA due to HIV infection, ART, or both, could exert long-term health effects in their children. 48 Studies to better understand and elucidate mechanisms underlying these changes will therefore also help to optimize the safety of ART regimens used during pregnancy. Several older studies found that HIV+, ART-naive persons had higher mtDNA-to-nDNA ratios than did ART-exposed persons, and that discontinuing ART (in particular, d-drugs with known mitochondrial toxicity) led to a statistically significant increase in this ratio. 49 –52 On the other hand, Maagaard et al. 2006 reported that mtDNA copies per cell in PBMCs did not differ between treatment-naive and NRTI-exposed persons, while individuals with HIV had significantly lower values than HIV-negative controls; lactate/pyruvate ratios were also higher in individuals with HIV. 7
HFE gene variants are much less prevalent in non-European-ancestry populations, suggesting another reason for the higher risk of ART-associated neuropathy (and possibly also other mitochondrial toxicities) among African Americans, which has been reported by some investigators. 53 However, it is important to note that despite the association with higher mtDNA content at baseline, HFE variant allele carriers also experienced greater declines in mtDNA copies per cell during the first 48 weeks of ART. Such effects suggest that HFE gene variants lead to either increased cellular mtDNA synthesis or increased numbers of mitochondria in PBMCs in the short term, which may protect against antiretroviral drug toxicity. However, these adaptations may not be sustainable during long-term ART and may ultimately augment mitochondrial stress and mtDNA depletion. Replication of these observations in other populations with HIV and further exploration of the mechanisms by which HFE exerts these effects are needed.
A limitation of our study is that we did not directly measure blood iron levels in study participants, although the published literature is relatively consistent in associating higher systemic iron stores, as reflected by levels of serum iron, transferrin saturation, and/or ferritin, with the presence of HFE variants. 54,55 A comparison group of otherwise similar individuals without HIV was also not available for mtDNA copy number measurements, so the individual contributions of HFE variants and HIV infection to mtDNA content in PBMCs could not be differentiated in this study. We did not include cumulative ddI exposure, or information about changes in d-drug exposure over the course of the study, as covariates in multivariable regression models, because this information was not available in a substantial proportion of individuals; similarly, data on cumulative exposure to all d-drugs were not available. Importantly, however, cumulative ddI exposure did not differ by HFE genotype, and we did adjust for ddI use at the time of blood sampling and for the total number of NRTIs, including d-drugs, that were used. Furthermore, in the primary analyses of these data, Simpson et al. (2006) found that neither d-drug antiretroviral use at baseline nor cumulative d-drug use was associated with total neuropathy score, arguing against a confounding effect by cumulative d-drug or NRTI use in this analysis. 22 Indeed, a growing number of genetic studies suggest that HIV infection, even in ART-naive individuals, may result in mitochondrial dysfunction and reduced cellular mtDNA content. 25 While no clear mechanism for this observation has yet emerged, our findings suggest that altered mtDNA biogenesis as a result of ART-induced disruption of cellular iron transport may contribute to this mitochondrial damage. 56
Additional studies are needed to investigate the role of HFE variants as well as systemic iron status and mtDNA copies per cell in mitochondrial toxicities of HIV infection and ART, in monitoring of disease severity during treatment, and in choice of treatment regimen. However, the findings reported in this study support the concept that iron metabolism and individual genetic variation in cellular iron transport significantly impact mtDNA levels and constitute one mechanism by which neurological complications in persons with HIV, due either to HIV disease itself or the effects of ART, may be influenced by host genetics.
Footnotes
Acknowledgments
We are deeply grateful to all study volunteers who participated in ACTG Study 384 and protocol A5117. We also wish to thank all of the ACTG clinical sites where participants were enrolled and followed, as well as the ACTG and the A5117 protocol team. Finally, we thank Cara Sutcliffe and Ping Mayo in the Vanderbilt Center for Human Genetics Research (DNA Core Laboratory) for genotyping assistance.
Funding support for these analyses was provided by NHLBI R21HL087726-03 (to AK), NIH 1P20GM113134 and 2 U54MD007601 (to MG), and NIH R21NS059330 (to TH).
The primary A5117 protocol was supported by the NIAID, AI38858, AI03855, AI25903, AI27664, AI046376, AI34853, AI27658, AI27670, AI46386, AI-25859, AI46370, AI32770, and AI27668, and the Neurologic AIDS Research Consortium funded by the National Institute of Neurologic Diseases and Stroke, NS032228, NS44807, and K24 NS002253 (DMS), and GCRC Units funded by the NCRR, RR00522, RR016467, RR00044, RR-00071, and RR00051.
Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number UM1 AI068634, UM1 AI068636, and UM1 AI106701. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.