
Abstract
Continual generation of HIV-1 recombinant forms might contribute to its genetic diversity. In this study, a novel B/C/CRF01_AE recombinant virus (GX2016EU13) was identified, which was isolated from a HIV-positive man who was infected through heterosexual sex in Guangxi, China. GX2016EU13 was identified as CCR5-tropic. As shown in the near full-length genome (NFLG) analyses, five recombinant breakpoints were found only in the pol gene of GX2016EU13, which divided the NFLG into three subtype B segments, two subtype C segments, and one CRF01_AE segment. The whole gag sequence of GX2016EU13 belongs to subtype B, and the whole env sequence of GX2016EU13 belongs to CRF01_AE. The recombinant form was distinct from the other circulating recombinant forms and unique recombinant forms reported. The emergence of GX2016EU13 may suppose the complexity of the HIV-1 epidemic in high-risk sexual populations in Guangxi, China.
Introduction
S
Guangxi, as an important traffic port in the southern of China, has much migrant population, which provides more opportunities to import new HIV-1 strains and increase transmissions. Now Guangxi province has become one of the most severe HIV-1 epidemic regions in China. 1,3 –5 Substantial second-generation recombinant forms have identified among high-risk populations, including intravenous drug users (IDUs), men who have sex with men and heterosexual population, since the first HIV infected case was reported in Guangxi in 1992. Most of HIV-1 subtypes prevalent in China could be found in there, including B, C, and CRF01_AE. 4 Besides, HIV-1 infection in Guangxi was spreading quickly from IDUs into the general population by sexual transmission, of which CRF01_AE has been identified as the dominant circulating recombinant form of HIV-1 in heterosexual transmission populations in Guangxi. 3,6 HIV-1 CRF01_AE subtype was originated from Central Africa and then was introduced into China in 1990s as the first identified CRF of HIV-1. 7 Subtype B strains were first identified in China among the IDUs in Yunnan province in 1989 and then became the most widespread HIV-1 strain in mainland China in terms of geographic reach. 8 Subtype C strains were also first identified in China among IDUs in Yunnan province in 1992. 9
In this study, the novel virus (GX2016EU13) was isolated from the sample collected from a 47-year-old HIV-1 positive patient infected by heterosexual sex in Guangxi, China. The patient was diagnosed as HIV-1 positive in 2013 and presented with CD4+ T cell count of 432 cell/μL and viral load of 379,000 copies/mL on April 14, 2016. The near full-length genome (NFLG) showed that it consisted of six segments from B, C, and CRF_01AE. This study was reviewed by the Institution Research Ethics Community of Chinese Center for Disease Control and Prevention (No.201334), and the informed consent was obtained from study participant.
The viral genome was extracted from the replicable virus using QIAamp Viral RNA Mini Kit and then transcribed into cDNA using the Superscript III first-strand synthesis system (Invitrogen). The NFLG was amplified with the gradient diluted cDNA template and TaKaRa LA Taq DNA polymerase (TaKaRa, Dalian, China), using the same nested polymerase chain reaction (nested-PCR) amplification conditions in both rounds, as described previously. 10 The positive PCR products were purified using the QIAquick Gel Extraction Kit (QIAGEN, Germany) and sequenced. Then the sequences were spliced and assembled by Sequencher V4.10.1. We finally obtained a length of 8,925 bp (relative to the HXB2 nucleotide numbering system: positions 636 to 9604) of the NFLG sequence of GX2016EU13. The NFLG sequences of HIV-1 references which included various subtypes (A–D, F–H, J, K) and CRFs circulating in China were downloaded from the Los Alamos National Laboratory HIV Database. Nucleotide sequences were first aligned using Clustal W and then adjusted manually using BioEdit. Phylogenetic trees were constructed with MEGA 5.0 using the neighbor-joining method with the Kimura two-parameter model and 1,000 bootstrap replications. Phylogenetic analysis suggested that the NFLG sequence of GX2016EU13 clustered with CRF01_AE, but formed a distinct monophyletic branch distantly related to CRF01_AE (Fig. 1). To determine the recombination form of GX2016EU13, the sequence was analyzed using RIP and SimPlot. Bootscan analysis was subsequently carried out to position the combination breakpoints with a window size of 200 bp, a step size of 20 bp, and 50% consensus. Analysis revealed that it was a unique mosaic structure composed of subtype B, C, and CRF01_AE (Fig. 2A). Five recombinant breakpoints were found in the pol gene of GX2016EU13 and divided the NFLG into six fragments: I B (636–2,540 nt), II C (2,541–3,034 nt), III B (3,035–3,177 nt), IV C (3,178–3,451 nt), V B (3,452–5,041 nt), and VI CRF01_AE (5,042–9,604 nt) (Fig. 2B, numbered according to the nucleotide sequence of HXB2, GenBank accession number K03455).
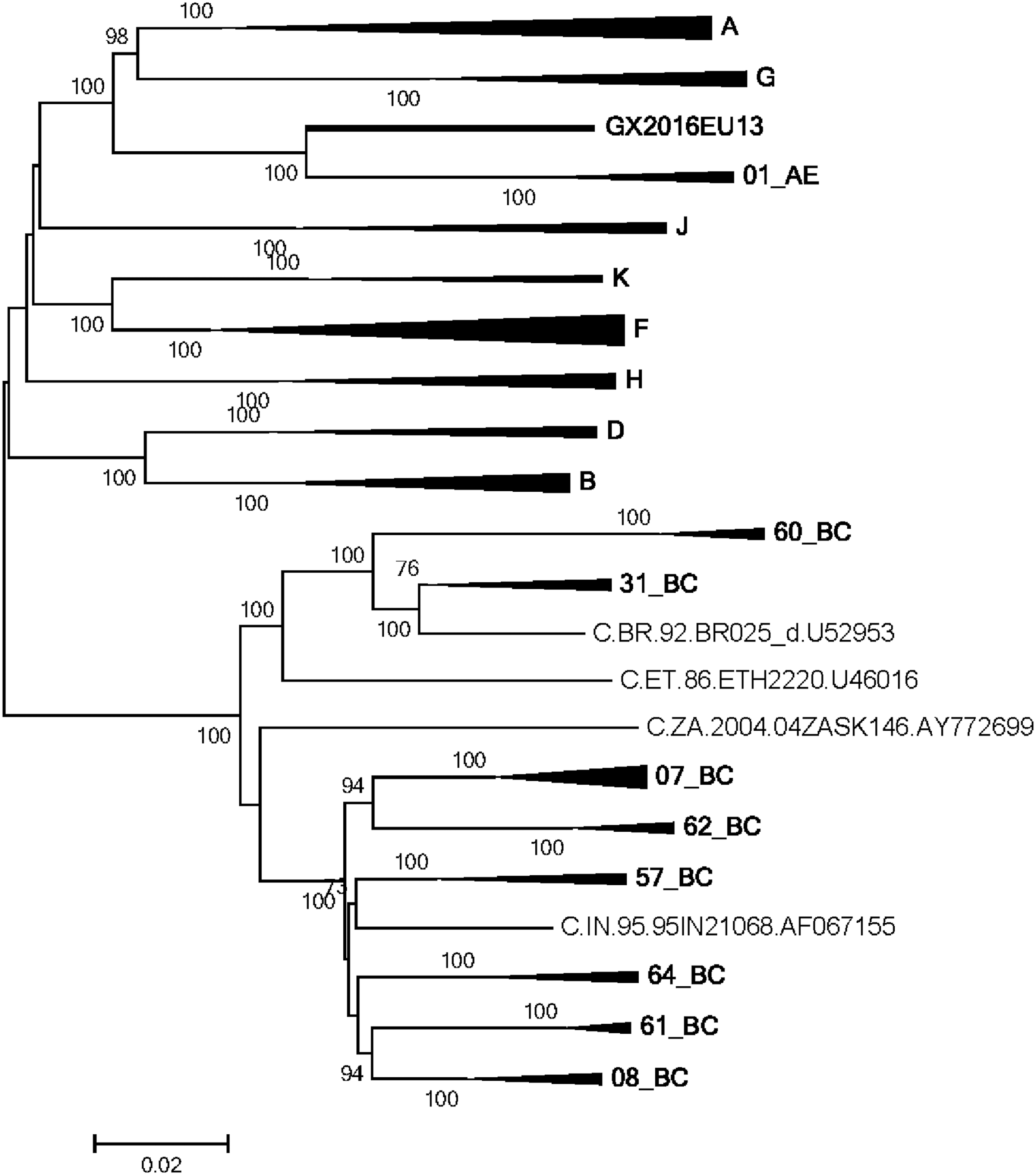
Phylogenetic tree analysis of the NFLG sequence of GX2016EU13. HIV-1 group M reference sequences were used to construct the neighbor-joining phylogenetic tree with the Kimura two-parameter model and 1,000 bootstrap replication test. The tree branch of GX2016EU13 is displayed as a bold line. Bootstrap values (>70) were showed at the corresponding nodes. The scale bar is shown at the bottom of the tree. NFLG, near full-length genome.
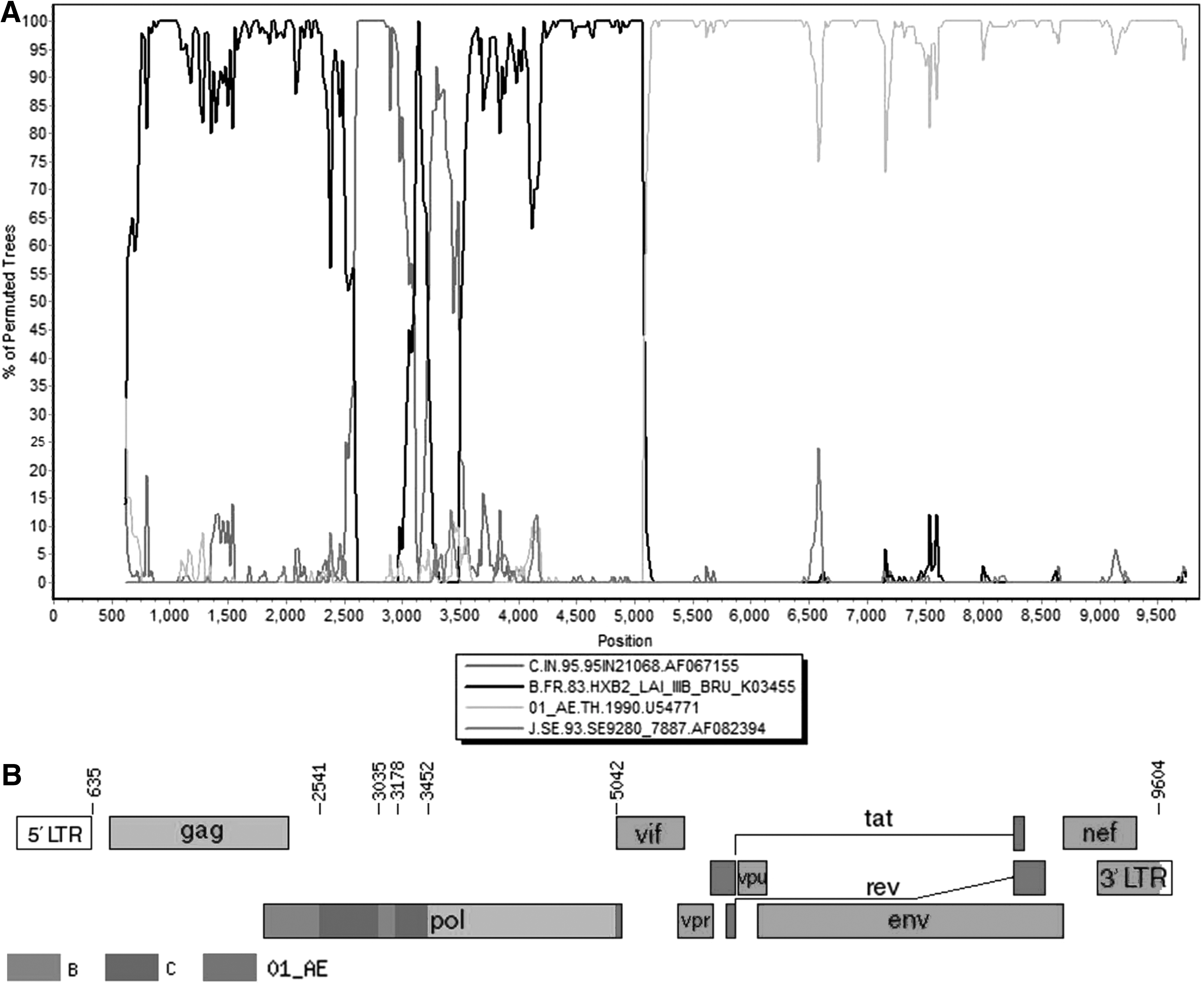
Recombinant analysis of the novel identified GX2016EU13.
To confirm the subtype of mosaic fragments, phylogenetic analysis for six genetic segments was carried out using the neighbor-joining method mentioned above (Fig. 3). Subregion tree analysis indicated that the parental origin of the subtype C segments was likely originated from the Indian subtype C lineage, which was also the parental origin of CRF07_BC and CRF08_BC. 2 In consequence of the shortness of C segments (II and IV), the bootstrap value of the reference strains was low. The CRF01_AE fragments VI CRF01_AE (5,042–9,604 nt) belonged to the CRF01_AE subcluster 2 lineage, which were found in the risk groups of Hetero and IDU populations. 11 The other regions of the genome (I, III, and IV) were most likely derived from subtype B, but we could not get enough signal for accurate inference of the evolutionary history of B segments. A potential reason for this could be that the parental origin of the B regions was from a variant of B that NFLG has not characterized. So the parental origins of the C, CRF01_AE, and B regions of the novel second-generation recombinant could be from Indian subtype C, CRF01_AE-2, and an unidentified B variant, respectively.
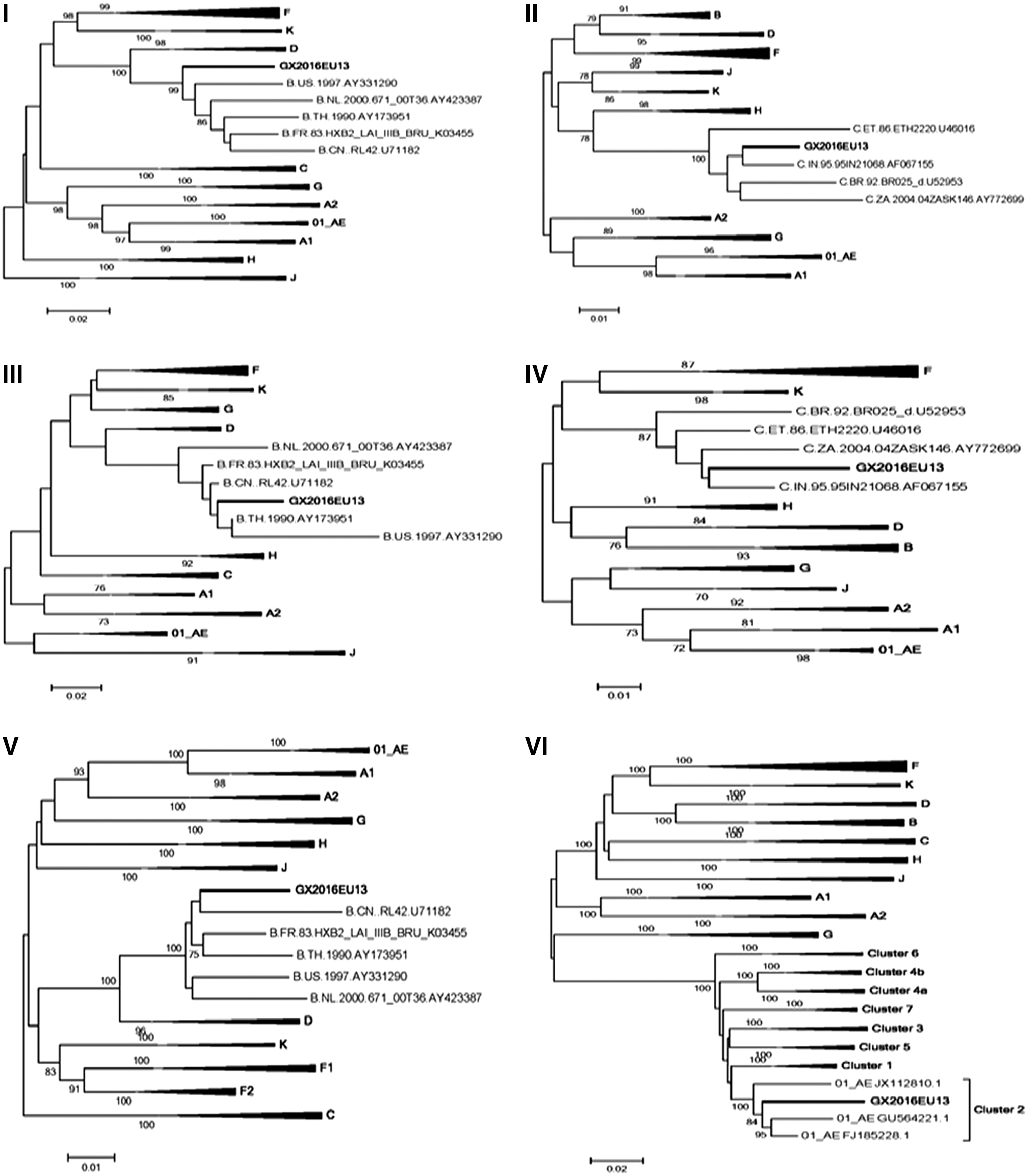
Subregion tree analysis of the novel identified GX2016EU13. The phylogenetic trees of the six mosaic segments (
In conclusion, GX2016EU13, isolated from a HIV-1 positive subject, was identified as a unique B/C/CRF01_AE recombinant form based on NFLG sequencing and as CCR-5 tropic on GHOST cells according to the method mentioned earlier. 10 The emergence of GX2016EU13 may reflect the continual generation of various forms of inter-subtype recombinants, which indicates the importance of continuous surveillance of the dynamic change of HIV-1 subtypes and recombinants in high-risk populations in Guangxi, China.
Sequence Data
The nucleotide sequence of the isolate GX2016EU13 has been submitted to GenBank with the accession number MG519330.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 81261120384, 81172733, and 81561128006); the European Research Infrastructures for Poverty Related Diseases (grant 312661); and the National Major Project of the State Key Laboratory of Infectious Diseases Prevention and Control (grant 2011SKLID102).
Author Disclosure Statement
No competing financial interests exist.