
Abstract
We investigated the relationship of monocytes, alveolar, and tissue-resident macrophage populations and the development of pulmonary arterial hypertension (PAH) in a nonhuman primate model of HIV infection. A prospective study of simian immunodeficiency virus-associated pulmonary arterial hypertension (SIV-PAH) was done. Rhesus macaques (n = 21) were infected with SIV. Blood, bronchoalveolar lavage fluid (BALF), and lung tissue were analyzed for monocyte and macrophage phenotypes and inflammatory mediators. Serial right heart catheterizations were performed at three time points throughout the study to assess hemodynamic alterations and the development of PAH. All 21 animals showed similar courses of SIV infection with an increasing proinflammatory plasma environment. At 6 months postinfection (mpi), 11 of 21 animals developed SIV-PAH (mPAP ≤25 mmHg; right ventricular systolic pressure [RVSP] ≤36 mmHg). PAH+ animals had an increased frequency of proinflammatory, nonclassical monocytes (CD14dimCD16+) (p = .06) in the peripheral blood and CD14+CCR7−CD163−CD206+ macrophages (p = .04) in BALF compared with PAH− animals at 6 mpi. Increased frequencies of these monocyte and macrophage phenotypes correlated with elevated RVSP (p = .04; p = .03). In addition, PAH+ animals had greater frequencies of tissue resident inflammatory M1-like CD68+STAT1+ (p = .001) and M2a-like CD68+STAT3+ macrophages (p = .003) and a lower frequency of anti-inflammatory M2c-like CD68+STAT6+ macrophages (p = .003) as well as fewer interleukin (IL)-10+ cells (p = .01). The results suggest that HIV-PAH is associated with skewing of monocytes and alveolar macrophages toward a proinflammatory, profibrotic phenotype. Furthermore, PAH+ animals may have diminished capacity to downregulate exaggerated chronic inflammation, as indicated by lower levels of IL-10 in PAH+ animals, contributing to disease progression.
Introduction
P
The prevalence of PAH is higher among HIV-infected individuals (0.4%–7%) compared with idiopathic PAH in the general population (15–50 cases per million people), although recent reports suggest that HIV-PAH may be more common than previously reported. 3 –7 Mechanisms promoting the increased prevalence and pathogenesis of HIV-PAH remain unclear, but growing evidence suggests that inflammation and aberrant immunologic responses contribute to idiopathic, 8,9 autoimmune, 10 and HIV-PAH. 11,12
This is supported by the observations of infiltration of inflammatory cells, including macrophages, lymphocytes, and dendritic cells (DCs) in pulmonary perivascular spaces 13 –15 and elevated levels of inflammatory markers, including interleukin (IL)-1β, IL-6, 16,17 MCP-1/CCL2, RANTES/CCL5, CX3CL1, 18 –20 platelet-derived growth factor, epidermal growth factor, 21 and vascular endothelial growth factor 22,23 in experimental models of PAH and in clinical studies. These factors can affect pathways influencing the proliferation, survival, and migration of vascular cells, including endothelial cells and pulmonary artery smooth muscle cells, suggesting that they may have a role in vascular remodeling associated with PAH. 24 –26
Monocytes and macrophages are key sources of inflammatory mediators, and their activation has been suggested to play a mechanistic role in PAH. 13,27 –29 Highly heterogeneous monocytes contribute to inflammatory processes, and subpopulations can be distinguished based on surface markers and biological functions. While classically activated CD14+CD16− monocytes exhibit phagocytic functions, alternatively activated, nonclassical CD14dimCD16+ monocytes patrol the endothelium and drive proinflammatory responses. 30,31 In response to different stimuli, monocytes traffic into tissues to differentiate into macrophages and DCs, which play critical roles in protective immunity and tissue homeostasis. 32
Within the lung, tissue resident and alveolar macrophages may contribute to pathological processes and their responses to increased or prolonged immune activation can cause significant tissue damage, fibrosis, and tissue remodeling. 33 –35
Macrophage plasticity includes a spectrum of functions related to inflammation, phagocytosis, and tissue repair, and these phenotypic and functional changes are expressed in response to environmental factors. 36,37 Macrophages are classified as M1 and M2 with further subclassification based on phenotypic markers and functionality. M1 macrophages (CD14+CCR7+CD163−CD206−) are highly microbicidal and are characterized by the secretion of inflammatory cytokines.
The M2 macrophage population is heterogenous and encompasses subpopulations M2a and M2c that can reversibly and progressively change the pattern of functions that they express. Because the phenotypic identity of these subpopulations is plastic and influenced by the local cytokine environment, the nomenclature is imprecise. It has been suggested that a more informative foundation for macrophage classification is based on macrophage function, including host defense, tissue repair, and immune regulation. 36,38
A shift toward alternatively activated monocytes has been observed in primary PH, systemic scleroderma PAH patients and has also been associated with endothelial dysfunction in patients with coronary artery disease. 39 –41 The accumulation of macrophages is a common feature of pulmonary vascular lesions in PAH patients and animal models of the disease. In the experimental model of hypoxia-induced PAH, a link between macrophage accumulation and M2 activation has been described, and an excessive polarization of wound-healing macrophages has been associated with tissue damage and fibrosis. 33 –35,42
Several studies have shown that SIV-infected macaques develop pulmonary vascular lesions and hemodynamic alterations that are characteristic of HIV-PAH. 15,43 –45 Despite the histologic and physiologic similarity of the SIV-PAH model to human PAH, the mechanisms underlying the development of HIV-PAH are not clearly understood. Likewise, the development of PAH cannot be predicted in HIV-infected individuals based on clinical parameters such as viral load (VL) or CD4 levels, 46,47 thus, a model of PAH progression would be highly informative in predicting disease progression and in evaluating treatment efficacy.
In a prospective study in rhesus macaques, we have shown that SIV-PAH developed between 6 and 12 months post-SIV infection in approximately 52% of the infected animals, however, we found no evidence that SIV infection severity, as measured by VL and CD4+ T cell levels, directly contributed to the susceptibility or severity of SIV-PAH. 48 Thus, we hypothesize that indirect effects, such as chronic inflammation, may promote the progression of SIV-PAH, and that monocytes and macrophages are central regulators of the pathogenic process. In this study, we investigated inflammatory mediators and phenotypic changes in monocytes and alveolar macrophages as driving forces or protective measures in SIV-PAH.
Materials and Methods
Animals
Twenty-one (9 female, 12 male) adult Chinese rhesus macaques (Macaca mulatta) were used and screened for simian retroviruses and other pathogens before purchase. Macaques were infected with SIVΔB670 (1:100 in PBS, tissue culture infectious dose of 50% (TCID50) = 2.6 × 10
5
, intravenously or mucosally. This virus induces CD4+ T cell decline and an AIDS-like disease (Supplement Fig. S1; Supplementary Data are available online at
All animal procedures were approved by the University of Pittsburgh Institution Animal Care and Use Committee. Research was conducted under the assurance number A3187-01 (Office of Laboratory Animal Welfare of the Public Health Service), following the guidelines described in the NIH Guide for the Care and Use of Laboratory Animals. 50 All animals were maintained in a Biosafety Level 2+ primate facility. The University of Pittsburgh is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Plasma, peripheral blood mononuclear cell, and total bronchoalveolar lavage fluid cell collection.
Peripheral blood and bronchoalveolar lavage fluid (BALF) were collected at baseline (BL), 6 months postinfection (mpi) and at study termination (10–12 mpi), as described previously. 51,52 In brief, plasma was isolated from 10 mL of EDTA-treated whole blood by centrifugation. Peripheral blood mononuclear cells were isolated treating whole blood with red blood cell lysis buffer. Plasma was stored at −80°C until further use and cells were either preserved in fetal bovine serum with 10% dimethyl sulfoxide (DMSO) until further analysis or directly used for flow cytometric analysis. Total fresh BALF cells were used for flow cytometric analysis, and differential cell counts performed was performed on Diff-quick (Siemens Health Care, Erlangen, Germany) stained cells. BAL fluid was stored at −80°C until further use.
Cytokine measurement in plasma and BALF
Quantitative analysis of cytokines, chemokines, and growth factors in plasma and BALF was performed using Cytokine 29-Plex Monkey Panel (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Plasma and BALF transforming growth factor-beta (TGF-β) and C-reactive protein (CRP) were quantified using TGF-β ELISA Kit: monkey (MyBioSource, San Diego, CA) and CRP (Monkey) ELISA Kit (Abnova, Walnut, CA) according to manufacturer's instructions. BALF samples were normalized based on the assumption that plasma and BALF have equal urea concentrations, 53 using QuantiChrom Urea Assay Kit (BioAssay Systems, Destin, FL) on plasma and BALF.
Flow cytometry
The following antibodies were used for the analysis of monocytes by flow cytometry: CD3 (clone SP34-2)–AF488, CD20 (clone 2H7)-BUV395, CD14 (clone MSE2)–APC, and CD16 (clone 3G8)–APCH7, all from BD Biosciences (San Jose, CA). The following antibodies were used to characterize macrophages: CD14 (clone M5E2)–BV421 and CD163 (clone GHI/61)–PerCPCy5.5, both from Sony (San Jose, CA); CD206 (clone 19.2)–APC and CCR7 (CD197) (clone 3D12)–PE-Cy7, both from BD Biosciences; and using isotype controls BV421-mouse IgG2a, APC-mouse IgG1, Pe-Cy7-rat IgG2a, and PerCPCy5.5-mouse IgG1, all from BD Biosciences. Raw cytometry data were acquired on a LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo analysis software (BD Biosciences).
All cytometry experiments analyzing monocytes included fluorescence-minus-one controls, and experiments analyzing macrophages included isotype controls. Doublet cells were excluded from analyses based on forward scatter-A and forward scatter-H. For each experiment, the monocyte and macrophage population was gated using forward and side scatter. For the characterization of monocytes, CD3- and CD20-positive cells were excluded, and the monocyte populations analyzed were based on their differential expression of CD14 and CD16. Monocyte phenotypes were defined as classical (CD14+CD16−) and nonclassical (CD14dimCD16+) 54 (Supplementary Fig. S2A). Cell numbers were calculated based on total monocyte counts and blood differentials. BL monocyte data were not available for one animal.
For the analysis of BALF macrophages, the CD14+ population was further characterized based on their expression of CCR7 (CD197), CD163, and CD206.
Alveolar macrophages were classified by flow cytometric analyses as M1-like (CD14+CCR7+CD163−CD206−) based on the expression of CCR7. 55 M2 macrophages (CD14+CCR7−) were further subclassified based on the differential expression of CD163 and CD206, (CD14+CCR7−CD163−CD206+) and (CD14+CCR7−CD163+CD206−) (Supplementary Fig. S2B). While CD206 has been identified with tissue repair and wound healing processes, 56 CD163 was associated with the resolving phase of this process. 57
Cell numbers were calculated based on the volume of retrieved BALF during bronchoscopy and macrophage counts using a hemocytometer. BALF were identified and quantified by differential staining (Diff-Quik) of total BAL cells and generally constituted less than 2%. Macrophage data were not available for three animals at BL, one at 6 mpi, and two at the terminal time point.
Right heart catheterization
Right heart catheterizations were performed on sedated animals at BL, 6 mpi, and before study termination (10–12 mpi), as previously described 15,48 using a Swan-Ganz balloon wedge pressure catheter advanced through the right atrium, right ventricle, and the pulmonary artery. Pressures were recorded with a Biopac M100 Acquisition system (Biopac, Goleta, CA). Waveforms were evaluated and mean values [right ventricular systolic pressure (RVSP), right ventricular diastolic pressure, right ventricular end systolic pressure, right atrial pressure, and pulmonary arterial pressure (PAP)] were obtained from 3 to 5 recorded beats per parameter. In instances for which we were unable to directly access pulmonary artery, mPAP was calculated as described by Peacock and colleagues 58 and validated for rhesus macaques. 48
Data were not obtained for one animal at 6 mpi and two at study termination.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections (5 μm thick) were examined by immunohistochemical staining using the following antibodies: rabbit anti-pSTAT1 IgG (clone D3B7, 1:1,000; Cell Signaling Technology, Danvers, MA), mouse anti-pSTAT3 IgG (clone M9C6, 1:200; Cell Signaling Technology), rabbit anti-pSTAT6 IgG (phosphor Y641, 1:100; Abcam, Cambridge, MA), goat anti-rabbit IgG (3.0 × 10–4 mg/mL; Jackson ImmunoResearch, West Grove, PA), goat anti-mouse IgG (5.5 × 10–4 mg/mL; Jackson ImmunoResearch), mouse anti-CD68 IgG (KP1, 2.0 × 10–5 mg/mL; Invitrogen), mouse IgG1 (0.01 mg/mL; BioLegend, San Diego, CA), rabbit IgG (0.04 mg/mL; BioLegend), and rabbit anti-IL-10 (Abcam).
Primary antibodies were developed using VECTASTAIN ABC-AP and VECTOR Blue Alkaline Phosphatase (AP) Substrate Kit (Vector Labs, Burlingame, CA) supplemented with levamisole (Vector Labs) and 10% Tween-20 or VECTASTAIN Elite ABC Kit and DAB Peroxidase (HRP) Substrate Kit (Vector Labs). Coverslips were mounted with VectaMount Permanent Mounting Medium (Vector Labs).
Anti-CD68 antibodies were used in conjunction with antibodies to phosphorylated STAT1, STAT3, and STAT6 to distinguish between three different macrophage populations. Tissue macrophages expressing STAT1 are associated with inflammation 59 ; while STAT3 signaling plays a role in wound healing processes. In contrast, 60 STAT6 signaling is associated with reduction of inflammation. 61 M1-like macrophages were defined as CD68+STAT1+, M2a-like as CD68+STAT3+, and M2c-like as CD68+STAT6+. 62
Mouse IgG1 and rabbit IgG were used as double isotype controls, as well as in combination with individual primary antibodies. Additional hematoxylin and eosin staining of adjunctive slides was performed at the Department of Pathology, University of Georgia. Immunohistochemistry (IHC) images were acquired using a Olympus DP71 camera fitted to Olympus BX41microscope and Olympus cellSens Entry. For the quantification of IL-10, M1-like, M2a-like, and M2c-like macrophages, 20 fields were randomly selected and imaged at 20 × magnification. M2a-like (CD68+STAT3+) data are not available for seven animals.
Statistical analyses
All statistical analyses were performed using Prism (GraphPad, La Jolla, CA). Serial characterization of RVSP, inflammatory marker, monocytes, and macrophages phenotypes in PAH+ and PAH− macaques were analyzed using Mann–Whitney U and Wilcoxon signed-rank tests. To test for associations between RVSP and each immune marker, Spearman correlation was used. p-Values of less than .05 was considered significant.
Results
Description of cohort
SIV-PAH developed within 6–12 mpi in 11 out of 21 animals (Supplementary Table S1 and Supplementary Fig. S1). SIV-PAH was diagnosed using human hemodynamic parameters (mPAP ≤25 mmHg; RVSP ≤36 mmHg). 63 In addition, anatomic evidence of PAH was evaluated based on periarterial and ventricular collagen deposition, pulmonary artery lesions, and perivascular lymphocytic follicle formation. Full hemodynamic analyses of this cohort are reported elsewhere. 48
SIV-infection induces a proinflammatory plasma and BALF environment
We have previously shown that gender, VL, and CD4+ T cell levels are not directly associated with PAH in the non-human primate model (Supplementary Fig. S1) (Tarantelli et al. in press), 43 suggesting that indirect factors may contribute to SIV-PAH. We examined the association between inflammatory mediators and the PAH phenotype in this cohort. Plasma and BALF levels of 31 inflammatory mediators and growth factors were analyzed following SIV infection (Supplementary Table S2).
Several plasma proinflammatory cytokine levels were increased during the course of SIV infection, including IL-6 and TNF-α 48 but there was no difference in levels of these cytokines between PAH+ and PAH− animals. Likewise, monocyte-associated chemokines MCP-1/CCL2 (p = .0002), MDC/CCL22 (p = .03), and IP-10/CXCL10 (p = .002) increased during chronic SIV infection (Fig. 1A(1–3)), although none of these distinguished SIV-PAH+ from SIV-PAH− animals. While levels of MCP-1/CCL2 (p = .05) and IP-10/CXCL10 (p = .04) were already significantly increased at 6 mpi, increased levels of MDC/CCL22 were only observed later in SIV-infection.
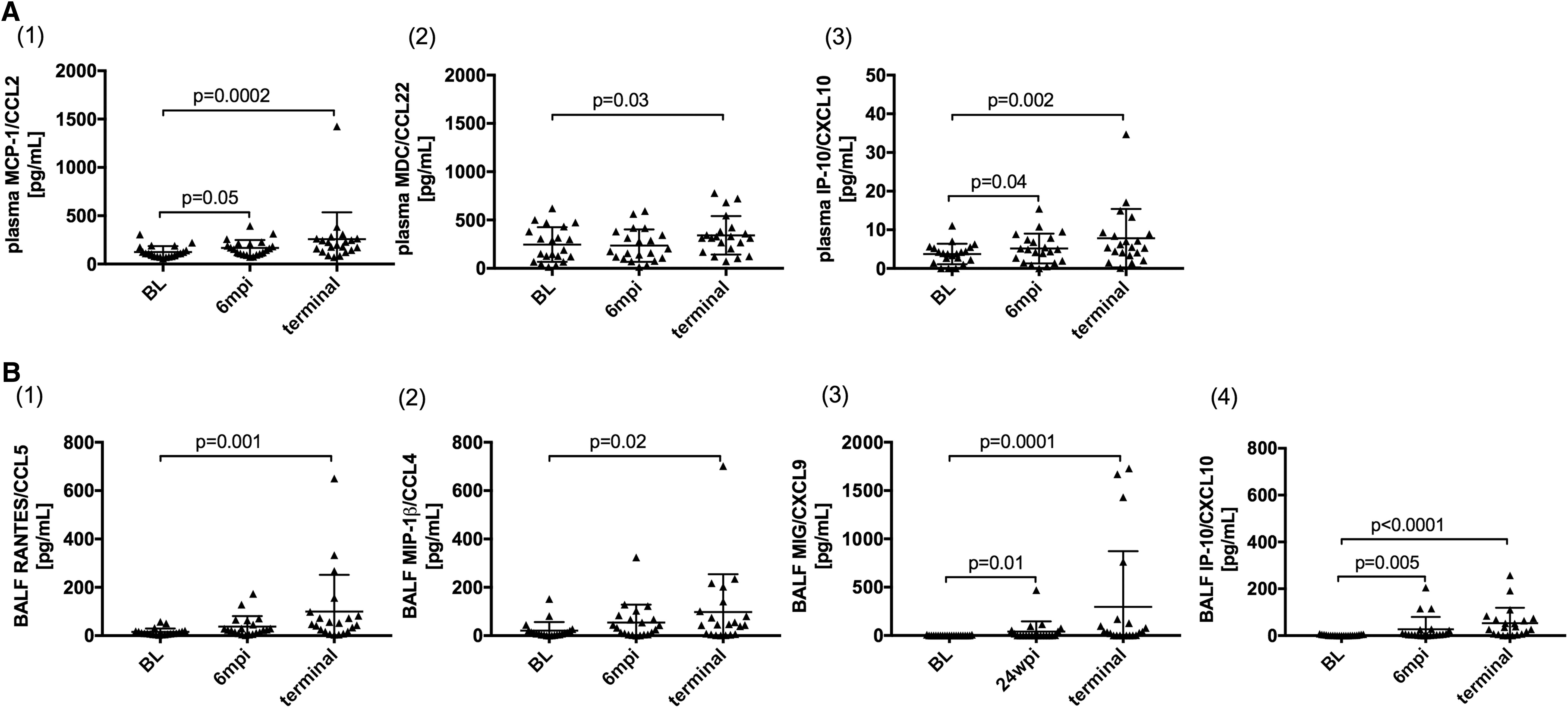
SIV-infection induces a proinflammatory plasma and BALF environment. Monocyte and macrophage-associated cytokines and chemokines were quantified in
In BALF, levels of M1-macrophage-associated chemokines, RANTES/CCL5 (p = .0005), MIP-1b/CCL4 (p = .02), MIG/CXCL9 (p = .0001), and IP-10/CXCL10 (p < .0001) increased in chronic SIV infection, of which MIG/CXCL9 (p = .01) and IP-10/CXCL10 (p = .005) were already increased at 6 mpi (Fig. 1B(1–4)). There was no difference between PAH− and PAH+ animals among these BALF chemokines.
Increased frequency of nonclassical monocytes correlates with elevated RVSP in SIV-PAH
The perivascular recruitment and accumulation of inflammatory cells, especially of the monocyte/macrophage lineage, has been reported to be associated with PAH. 13,27,28 In this study, the peripheral blood monocyte count, determined by blood differentials, remained unchanged from BL to terminal SIV infection and was not associated with PAH (Fig. 2A(1–3)).

Increased frequency of nonclassical monocytes correlates with elevated RVSP in SIV-PAH.
We examined the relationship of alterations in monocyte phenotypes to pulmonary artery pressure during the course of SIV-infection. The number of CD14dimCD16+ nonclassical monocytes remained unchanged over the course of SIV-infection (Fig. 2B(1)). However, PAH+ animals had more nonclassical monocytes (p = .06, Fig. 2B(2)) than PAH− at 6 mpi. The number of nonclassical monocytes also correlated with elevated RVSP (p = .04, R = 0.46, Fig. 2B(3)). CD14+CD16− classical monocytes increased at 6 mpi compared to BL in both PAH+ and PAH− animals (p = .03, Fig. 2C(1)).
There was no difference in the frequency of classical monocytes at any of the time points in PAH+ and PAH− (p = .97, Fig. 2C(2)), and there was no association between classical monocyte numbers and RVSP (Fig. 2C(3)). At the terminal time point, there was no significant difference in nonclassical monocytes between PAH− and PAH+ animals. At this late stage in SIV-infection, the frequency of CD14dimCD16+ monocytes increased in PAH− animals and remained unchanged in PAH+ animals. However, these changes in the monocyte compartment may be influenced by infections associated with end-stage AIDS (data not shown).
Alterations in BALF macrophage populations correlate with elevated RVSP in SIV-PAH
Patrolling monocytes respond to chemokines by trafficking into tissues, where they differentiate into macrophages and DCs. 32 Before SIV-infection, alveolar macrophages comprised approximately 80% of BALF cells; these decreased to approximately 50% in chronic SIV infection (p = .01, Fig. 3A(1)). At 6 mpi, PAH+ animals had higher overall frequencies of alveolar macrophages compared to PAH- (p = .01, Fig. 3A(2)). The M1-like population (CD14+CCR7+CD163−CD206−) significantly increased in chronic SIV infection (Fig. 3B(1)), and the M2 subpopulation (CD14+CCR7−CD163−CD206+) was increased at 6 mpi, compared to BL (Fig. 3C(1)). This M2 subpopulation significantly increased in PAH+ compared with PAH− (p = .04, Fig. 3C(2)) at the 6 mpi time point and correlated with elevated RVSP (p = .03, R = 0.5; Fig. 3C(3)).
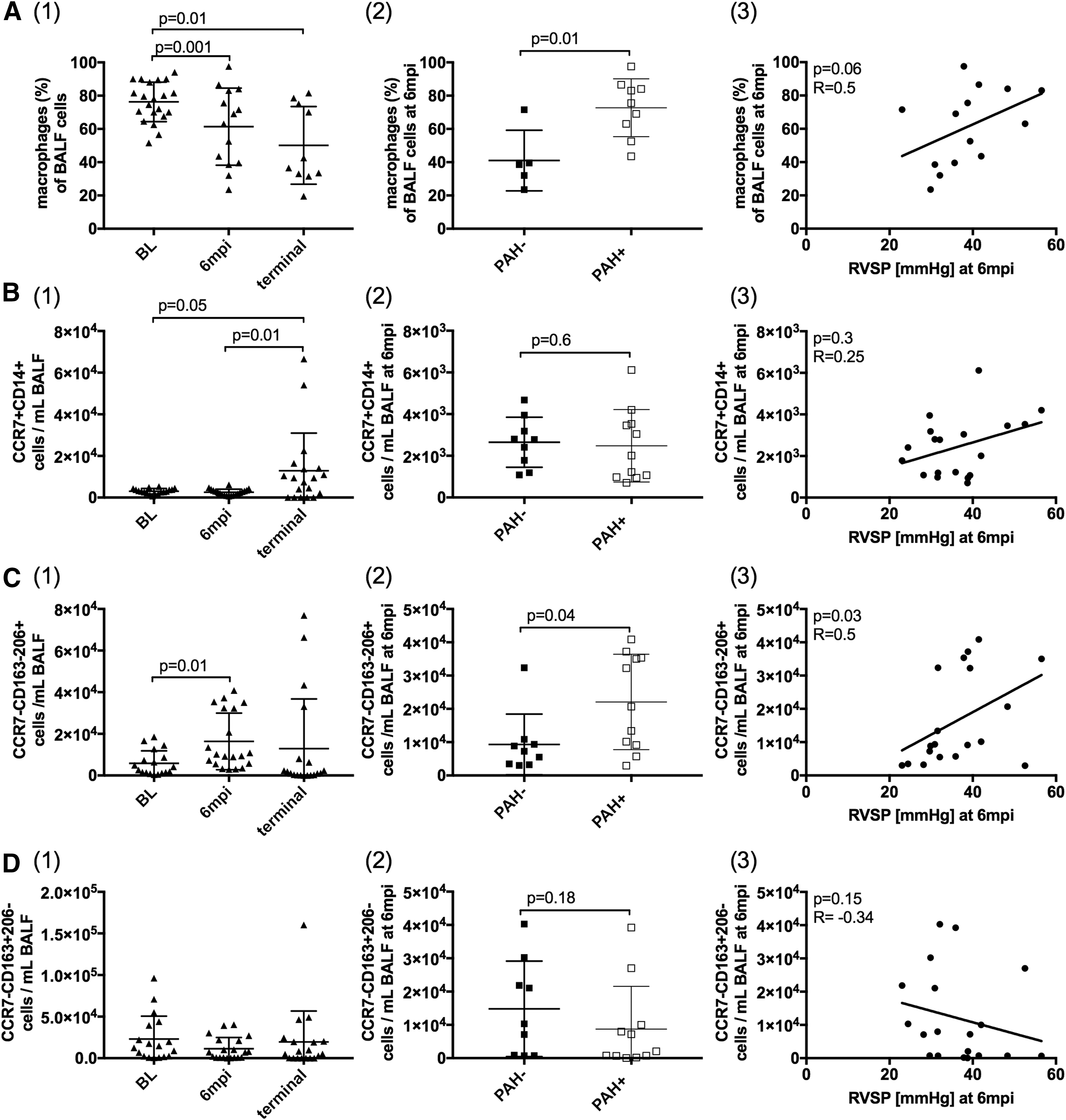
Alterations in BALF macrophage populations correlate with elevated RVSP in SIV-PAH.
The numbers of M2c-like CD14+CCR7−CD163+CD206− did not change significantly throughout infection compared to BL levels (Fig. 3D(1)). At the terminal time point, CD14+CCR7−CD163−CD206+ cell frequency decreased in all animals, resulting in only a minimal difference between PAH− and PAH+ groups. These changes in the macrophage compartment, however, may be confounded by opportunistic infections associated with end-stage AIDS (data not shown).
IHC evaluation of lung tissue
Characterization of macrophage phenotypes of BALF samples by flow cytometry captures mainly alveolar macrophages. To further examine the pulmonary tissue environment in the context of SIV infection and PAH, we analyzed tissue-resident macrophages by IHC and characterized populations based on STAT expression. 62 PAH+ animals had increased frequency of tissue resident, M1-like cells (CD68+STAT1+) (p = .001, Fig. 4A), and M2a-like cells (CD68+STAT3+) (p = .003, Fig. 4B), compared with PAH− (Supplementary Table S3). In contrast, PAH− animals had a higher frequency of M2c-like CD68+STAT6+ macrophages compared with PAH+ (p = .003, Fig. 4C and Supplementary Table S3).
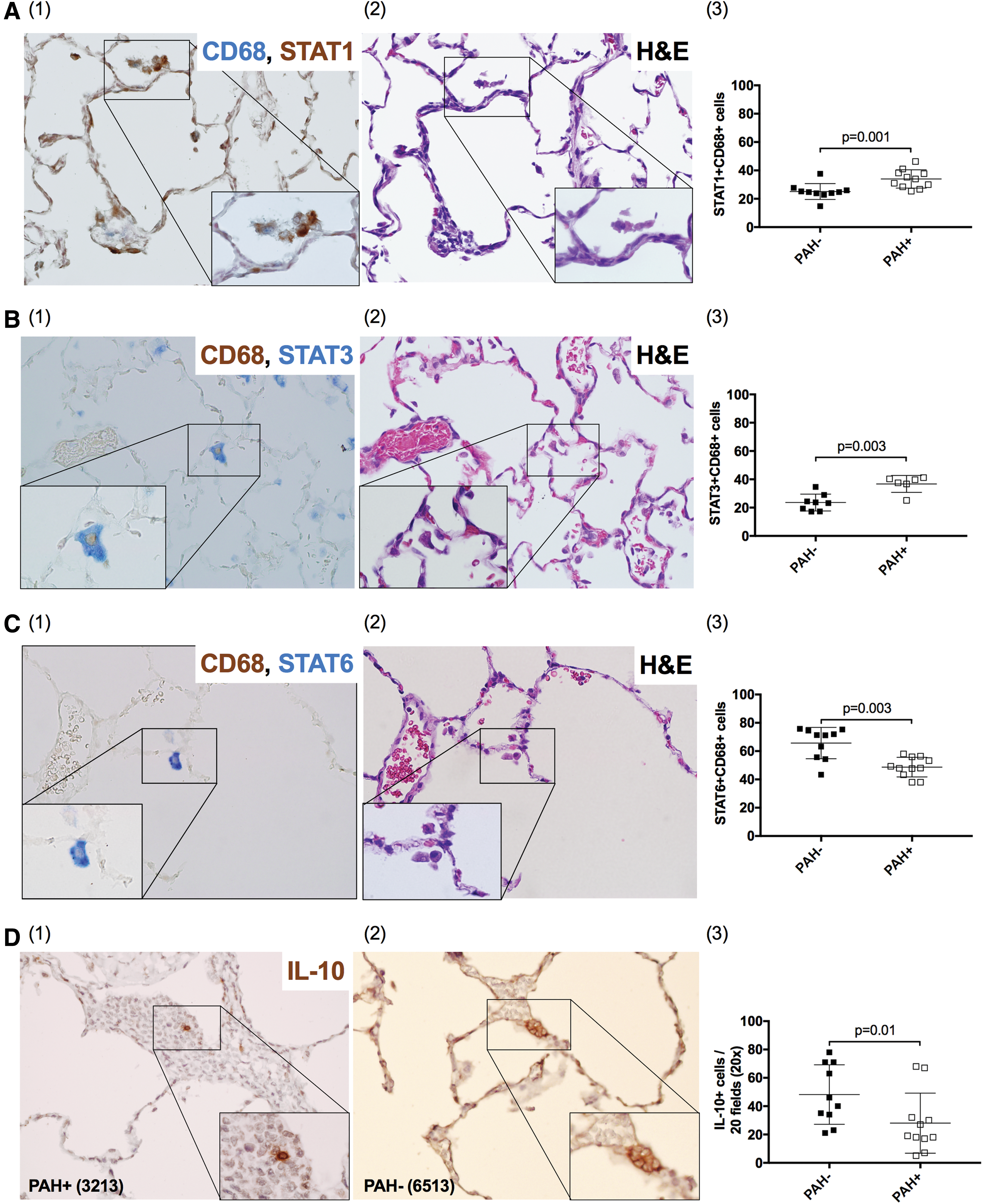
IHC evaluation of lung tissue. In addition to alveolar macrophages in BALF, tissue resident macrophages were characterized by the expression of CD68 and
Lung tissue was also evaluated for cells expressing the immunomodulatory cytokine IL-10. We found that PAH− animals showed higher numbers of IL-10+ expressing cells compared with PAH+ (p = .01, Fig. 4D and Supplementary Table S3).
Discussion
We investigated the relationship between inflammatory mediators, phenotypic changes in peripheral blood monocytes and pulmonary macrophages, and the development of PAH during experimental SIV infection. We identified increased frequencies of proinflammatory, nonclassical peripheral blood monocytes and M2 macrophages in BALF samples and pulmonary tissue in SIV-infected animals that developed PAH. The skewing of the monocyte/macrophage populations toward proinflammatory and profibrotic phenotypes correlated with increased pulmonary and right heart pressures. In contrast, we found that SIV-infected animals that maintained normal hemodynamic profiles throughout the study had increased frequencies of lung anti-inflammatory, M2c-like macrophages (CD68+STAT6+), and increased production of lung IL-10.
As expected, levels of multiple proinflammatory cytokines and chemokines increased during the course of SIV infection, including those associated with monocyte and M1-macrophage trafficking such as IL-1β, IL-6, TNF-α, MCP-1/CCL2, MDC/CCL22, IP-10/CXCL10, RANTES/CCL5, MIP-1β/CCL4, and MIG/CXCL9. Interestingly, none of these factors directly associated with SIV-PAH.
Cells of the monocyte/macrophage lineage are highly heterogeneous with respect to functionality and plasticity, responding to environmental stimuli, and activation is associated with changes in functional phenotypes. 36,38 The phenotype of polarized macrophages is reversible to some extent, thus definitive phenotypic characterization is challenging in the in vivo environment. Pathology can be associated with changes in macrophage polarization, with classically activated M1 cells promoting inflammation and M2 or M2-like cells associated with tissue repair, wound healing, or chronic inflammation. 64
As macrophages polarization has been reported to be associated with PAH 42 and these cells are central regulator of the inflammatory response, 33 –35 we sought to characterize the influence of SIV infection on alveolar macrophages and tissue resident pulmonary macrophages and their relationship to the development of PAH. In BALF, M1-like macrophages were defined as CD14+CCR7+CD163−CD206−, and two different M2 subpopulations CD14+CCR7−CD163−CD206+ and CD14+CCR7−CD163+CD206− were distinguished in the context of SIV-associated PAH. The chemokine receptor, CCR7 (C–C chemokine receptor type 7) has been shown to be upregulated in M1 polarized macrophages and is induced by LPS and IFN-γ. 55 M2 macrophages are characterized by the downregulation of CCR7 and the differential expression of several markers, including CD163 and CD206.
Mirza and Koh 56 found upregulated CD206 in tissue repair processes in mice, and CD163 expression was shown to be increased at a later stages during the resolution of wound healing. 57 In the present study, we found that increased frequencies of CD14+CCR7−CD163−CD206+ in PAH+, which correlated with elevated RVSP.
We further evaluated tissue macrophages based on CD68 and the phosphorylation of STAT1, STAT3, and STAT6 to distinguish between the three different macrophage subpopulations. 62 M1-like macrophages were defined as CD68+STAT1+. IFN-γ has been shown to induce phosphorylation of STAT1, which in turn promotes inflammation, including inducing expression of chemokines, regulating the differentiation and death of hematopoietic cells, and promoting production of reactive oxygen species and nitric oxide. 59
M2a-like macrophages were defined as CD68+STAT3+, as STAT3 signaling plays a crucial role in normal wound healing with target genes, including HGF and VEGF, and response to injury. 60,65 M2c-like macrophages were identified as CD68+STAT6+, since STAT6 signaling has been shown to reduce inflammation by suppressing NF-kappa B transcriptional activation. 61,66 In SIV-infected macaques that developed PAH, we found increased frequencies of CD68+STAT1+ and CD68+STAT3+ macrophages, while the M2c-like CD68+STAT6+ population was decreased. This scenario of increased frequency of proinflammatory, profibrotic macrophages populations in the lung tissue and a decrease in the frequency of downregulatory cells is consistent with the inflammation-mediated pathogenesis of PAH.
Despite the fact that antiretroviral therapy has transformed HIV infection from a highly immunosuppressive disease to a chronic disease state, HIV-PAH remains a severe complication with no effective treatment or cure. The prevalence of HIV-PAH has been estimated at 0.4%–7%. This estimate likely underrepresents the actual frequency, as recent studies have reported elevated RV pressures in 35%–57% of HIV-infected patients by echocardiography (35%–66%> 30 mmHg; 6.6%–15.5%> 40 mmHg). 3 –7 Mechanisms promoting the increased prevalence and poorer prognosis of HIV-PAH remain unclear. Evidence, including the work we present here, supports a role for chronic immune activation and inflammation in HIV infection with the development of HIV-associated comorbidities, including PAH. 11,12
PAH is diagnosed at all stages of HIV infection with no consensus on the relationship among CD4+ T cell counts, VL, and PAH. 46,47 In this model, clinical parameters, including peak VL, viral set points or plasma CD4+ T cell nadir were not predictive of the development or severity of SIV-PAH. 15,48 These findings do not exclude the possibility that specific viral proteins play a role in the pathogenesis; however, the lack of an association among VL, level of inflammation, and incidence or severity of HIV-PAH supports the hypothesis that secondary factors such as genetic factors or host-specific responses to viral infection may alter the disease progression.
SIV and SIV-HIV chimeric virus infections of susceptible nonhuman primate species are well characterized models of human HIV infection with hemodynamic alterations and histopathologic evidence of naturally occurring PAH 15,44,45 and provides a means for investigating the pathogenesis of HIV-PAH in a highly relevant animal model. We have reported that in the absence of potential confounding parameters of HIV-PAH studies such as antiretroviral use, preexisting lung disease, cigarette smoking, and intravenous drug use, SIV infection alone led to PAH in ∼52% of animals within 6–12 months of infection, while the remaining did not show changes in their hemodynamics.
The present study provides insight into the relationship between chronic immune activation, particularly in the monocyte/macrophage compartment and PAH pathogenesis in the SIV model of HIV infection. Phenotypic changes in peripheral blood monocytes have been described during HIV as well as experimental SIV infection in several macaque models. HIV infection and other inflammatory conditions have been associated with an expansion of CD16+ nonclassical monocytes. 67 –69 Nonclassical monocytes have also been associated with wound healing processes and coronary heart disease. 31,70 While we did not see an overall increase in the nonclassical monocyte population during the course of infection, we detected higher frequencies of this phenotype in PAH+ animals. This increased frequency of nonclassical monocytes significantly correlated with elevated pulmonary arterial and right heart pressures.
Depending on the stimuli, monocytes differentiate into M1 or M2 macrophage phenotypes promoting either inflammation, tissue homeostasis or repair. 32 During acute HIV infection, M1 activation is believed to be induced by high levels of proinflammatory cytokines. 71,72 This activation is crucial for promoting an effective immune response, however, continued stimulation of macrophages can lead to immune dysfunction, tissue damage, and disease pathology 33 –35 that may include PAH.
At later stages of HIV infection, a phenotypic shift of M1 toward M2 has been reported which parallels the HIV-associated Th1/Th2 switch. 35,73 Within the population of M2 macrophages, dysregulated M2a responses, which promotes the resolution of injury and wound repair, are associated with extensive tissue remodeling and fibrosis. 74,75 In our study, we found the increased M2 subpopulation (CD14+CCR7−CD163−CD206+) in BALF of PAH+ animals at 6 mpi. As BALF samples represent primarily alveolar macrophages, we further evaluated the pulmonary immune responses by analysis of tissue-resident macrophages, which may provide additional insight into local regulation of tissue inflammation and remodeling. In lung tissue of PAH+ animals, we found increased frequencies of M1-like and M2a-like macrophages as well as a decreased frequency of M2c-like.
M2c macrophages are stimulated by IL-10, which can be protective in lungs in chronic viral infection models and subsequent tissue remodeling. 76,77 Several studies describe a central regulatory role of IL-10 in limiting exaggerated inflammatory responses, preventing damage to the host and maintaining normal tissue homeostasis. Studies in IL-10-deficient mice revealed that they were able to clear pathogens more efficiently but this response was often accompanied by significant immunopathology. 78,79 A study analyzing dysregulated IL-10 signaling in the intestinal mucosa of SIV-infected rhesus–macaques described epithelial cell damage as a contributing factor to subsequent SIV pathogenesis. 80
In the present study, we found reduced expression of IL-10 in PAH+ animals, compared with PAH− in the lung, supporting the concept that a diminished capacity to downregulate chronic inflammatory processes may be part of SIV-PAH. Although the local source of IL-10 in these lung tissues was not determined, other studies have suggested that a deficiency in regulatory T cells and altered production of regulatory chemokines and cytokines, including IL-10, might contribute to pulmonary vascular dysfunction. 81 –83
There are limitations to our study's ability to discern mechanisms underlying PAH. Due to the relatively small study size, we may have lacked sufficient power to detect associations between inflammatory mediators and development of PAH. Nevertheless, despite a generalized, proinflammatory environment associated with SIV infection, we have identified a link between the development PAH and the alternatively activated phenotype in the monocyte/macrophage compartment. Another limitation of this study is that we have not yet determined upstream parameters that drive the monocyte/macrophage skewing following SIV infection. This important question may be addressed by serial examination of lung tissue inflammation at earlier stages of disease progression.
In summary, this prospective study is the first to identify specific phenotypic changes in circulating and pulmonary monocyte and macrophage populations associated with PAH in a primate model of HIV infection. Although a generalized, systemic inflammation in HIV infection has been well characterized and associated with comorbidities, the results presented here begin to define specific proinflammatory, profibrotic cell populations that are associated with progression of SIV-PAH at early stages of disease. These are important findings that may contribute to the identification of early targets of intervention before the development of the irreversible pulmonary vascular remodeling associated with PAH.
Footnotes
Acknowledgments
The authors thank Dr. Christopher Janssen and Michael Bennett Johnston for veterinary support and Dr. Michael Murphey-Corb for providing SIV virus stocks and titration information. This work was funded by the National Institute of Health, National Heart, Lung, and Blood Institute (R01 HL138437, R01 HL131449) and the Georgia Research Alliance, Charles H. Wheatley Endowment (K.A.N.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.