
Abstract
Interferon-α (IFN-α) plays a vital role in combating viral infections especially in the early control after infection. However, the HIV infection has shown substantial level of suppression of IFN-α secretion during initial phase of infection. The reasons behind this impairment are still obscure. As plasmacytoid dendritic cells (pDCs) are the major producers of this cytokine, the mechanisms of HIV-1-mediated suppression of IFN-α production by pDCs using the primary pDCs were explored. The nuclear translocation of the interferon regulatory factor (IRF)-7, a transcription factor for IFN-α genes, is essential for the initiation of IFN-α production in pDCs. The HIV-1-exposed pDCs did not show the translocation of IRF-7 into the nucleus in our experiments. Furthermore, it was also observed that HIV-1 inhibited AKT phosphorylation of PI3K/akt pathway in pDCs, an important step for IRF-7 translocation to nucleus. HIV-1-induced inhibition of AKT phosphorylation and IRF-7 translocation was evident even in the presence of Toll-like receptor-7 agonist stimulation and correlated with IFN-α suppression. The findings suggest that HIV-1 may alter AKT phosphorylation to inhibit the translocation of IRF-7 into pDC nucleus, leading to IFN-α suppression, and this may be the reason for IFN-α abrogation observed in recently infected HIV patients. Understanding of interactions between HIV-1 and signaling pathways leading to IFN-α secretion may provide targets for immune intervention.
Introduction
T
IFN-α induction by pDCs upon HIV-1 exposure involves sensing of HIV-1 single-stranded RNA through endosomally located Toll-like receptor (TLR)-7. This recognition leads to cell signaling cascade involving molecules such as MyD88, IRAK1, IRAK4, and TRAF6, which results in the activation of interferon regulatory factor (IRF)-7 protein. The activated IRF-7 then translocates from cytoplasm to nucleus and activates residing IFN-α genes. Guiducci et al. 6 have reported that the activation of PI3K/akt pathway is a prerequisite for the translocation of IRF-7 from the cytoplasm to nucleus in pDCs. It suggests that PI3K/akt-driven IRF-7 translocation is critical for IFN-α production after HIV-1 recognition by pDCs. Hence, the role of HIV-1 in influencing PI3K/akt-driven IRF-7 translocation and suppression of IFN-α production in pDCs was explored in the present study.
Materials and Methods
Virus stock, pDC preparation, and stimulation with HIV-1 or TLR-7 agonist
HIV-1 (subtype C, isolated from a patient enrolled within 100 days of infection) stock was prepared as described earlier, 7 ultracentrifuged, filtered through 0.22 μm filter and used for in vitro stimulation of pDC cultures.
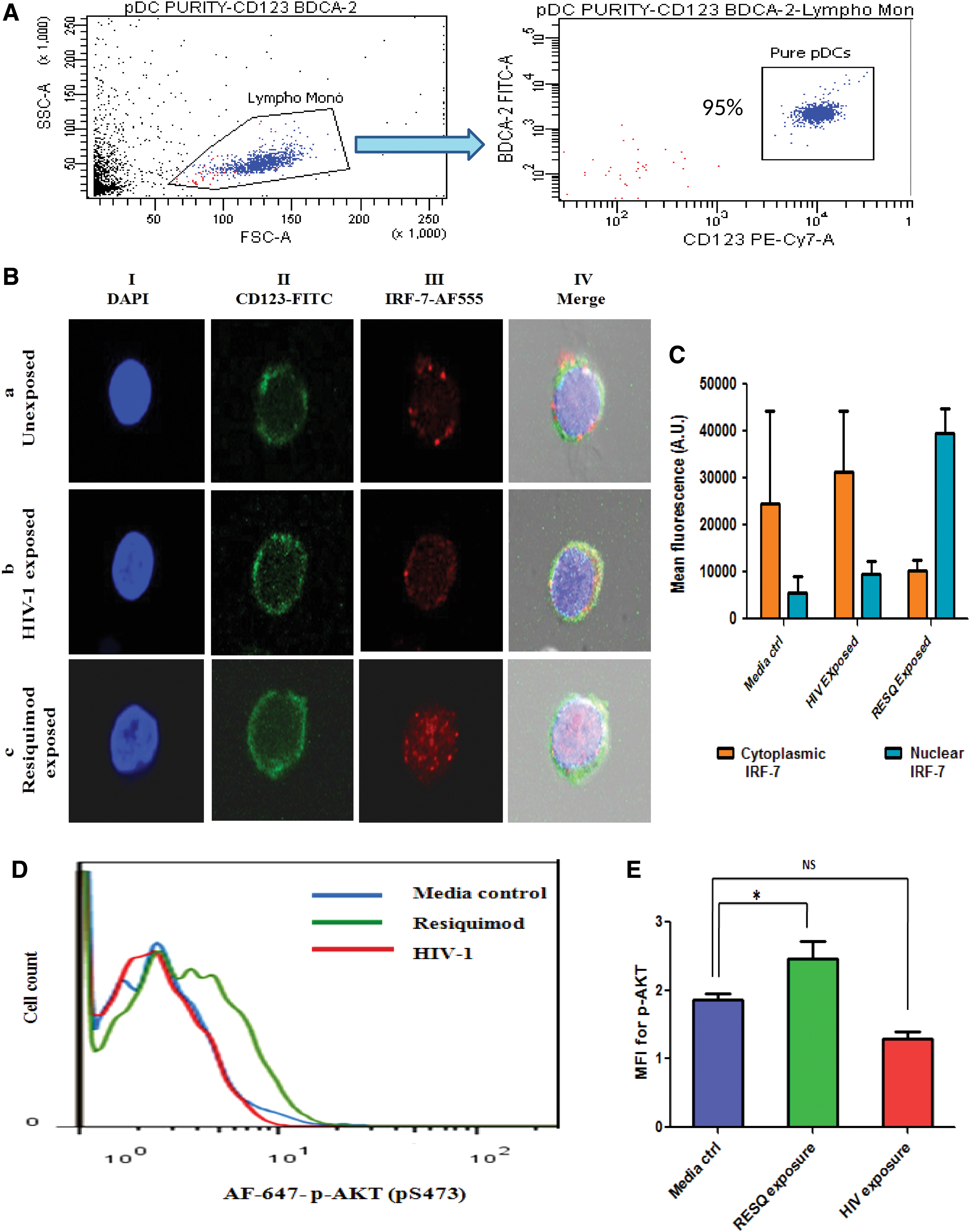
pDCs were isolated from healthy donor's blood or from buffy coats (source: a local blood bank) by negative selection using pDC isolation kit II, human (Miltenyi Biotec, Germany), with purity (CD123+BDCA-2+) ranging between 85% and 99% (Fig. 1A).
Purified pDCs were kept as 2 × 10
4
cells/100 μL of RPMI 1640 (Gibco-Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (Moregate Biotech, Bulimba QLD, Australia),
Confocal microscopy for IRF-7 localization
For studying the effect of exposure of pDCs to HIV on IRF-7 translocation, pure pDCs were exposed to Resiquimod or HIV-1 or left unexposed. In later experiment, pDCs were exposed to either Resiquimod or HIV-1 alone or were exposed to Resiquimod after pre-exposure to HIV-1 for 2 h. pDCs left unexposed were treated as negative control. All cultures were maintained at 37°C in 5% CO2 atmosphere. After 3 h, pDCs were fixed with 2% formaldehyde solution (15 min at 37°C) and stained for CD123 (CD123-FITC; Miltenyi Biotec) surface marker. Subsequently, cells were permeabilized with 100% methanol for 10 min at −20°C, blocked with 10% normal goat serum [45 min at room temperature (RT)], and further stained with anti-IRF-7 rabbit polyclonal antibody (Santa Cruz Biotechnology, Dallas, TX) followed by anti-rabbit IgG-AF555 (Invitrogen) (both at 1:100 concentration for 1 h at RT). Cells were then stained with DAPI (Sigma-Aldrich), cytocentrifuged (Shandon Cytospin II; ThermoFisher Scientific, Waltham, MA), and mounted on glass slides with Diamond antifade mounting medium (Invitrogen). Images were acquired on confocal microscope (LSM 510 META; Zeiss, Oberkochen, Germany) at 63 × magnification. The IRF-7 fluorescence in cytoplasmic as well as nuclear compartments from images was measured using FIJI (ImageJ V 2.0.0).
Flow cytometry for detection of phosphorylation of AKT
The pDCs were studied for AKT phosphorylation upon exposure to HIV-1. In these experiments, pure pDCs exposed to (1) Resiquimod or (2) HIV-1 or (3) Resiquimod and HIV-1 simulteneously or (4) Resiquimod after 2 h pre-exposure to HIV-1 or (5) medium control at 37°C in 5% CO2 atmosphere were immediately fixed with Fixation and Permeabilization solution (BD Biosciences) and surface stained with CD123-PE (BD Biosciences, San Jose, CA) and BDCA-2-FITC (Miltenyi Biotec) antibodies. Cells were further stained with anti-phospho-AKT-AF647 (BD Biosciences) antibody for 45 min at RT and acquired on flow cytometer (BD FACS Aria or FACS Calibur). 10 3 CD123+BDCA-2+ pDCs were considered for further analysis of phosphorylation of AKT using FlowJo software.
Enzyme-Linked Immunosorbent Assay for quantification of IFN-α
pDCs were exposed to HIV-1 or Resiquimod or Resiquimod and HIV or Resiquimod after 2 h pre-exposure to HIV-1 for 8 h, and supernatants were stored at −20°C for the measurement of IFN-α levels using IFN-α pan-specific enzyme-linked immunosorbent assay (ELISA) Kit (MabTech, Nacka Strand, Sweden) according to the manufacturer's instructions. Statistical significance in IFN-α levels between different exposures was calculated by the Mann–Whitney test.
Ethics statement
This study was carried out in accordance with the recommendations of Institutional Ethics Committee of the National AIDS Research Institute (NARI), Pune, India (NARI/EC Protocol No.: 2012-19). Blood samples were collected from HIV-negative healthy donors after taking their written informed consent.
Results
Exposure of pDCs to HIV-1 does not induce IRF-7 translocation or AKT phosphorylation
Nuclear translocation of IRF-7, a transcription factor for IFN-α gene induction, was studied in pDCs upon exposure to HIV-1. Pure pDCs (2 × 10 4 /100 μL pDC culture media) isolated from five healthy donors were exposed to 0.5 MOI of HIV-1 for 3 h, keeping Resiquimod (TLR-7 agonist) and unexposed pDCs as positive and negative controls, respectively. The exposure time was decided on the basis of earlier study. 6 The representative confocal microscopic images illustrate the cytoplasmic localization of IRF-7 [Fig. 1B-a.I–IV, C; mean fluorescence of 24,469 arbitrary units (AU) in the cytoplasm and 5463 AU in nuclear compartment] in unexposed pDCs and the nuclear translocation of IRF-7 due to Resiquimod exposure (Fig. 1B-c.I–IV, C; mean fluorescence of 10,288 AU in the cytoplasm and 39,431 AU in the nuclear compartment). HIV exposure did not lead to IRF-7 translocation, and IRF-7 was detected in the cytoplasm only (Fig. 1B-b.I–IV, C; mean fluorescence of 31,408 AU in the cytoplasm and 9547 AU in the nuclear compartment). These findings indicate that HIV-1 may play a role in the inhibition of translocation of IRF-7.
Considering the essentiality of PI3K/akt pathway for the translocation of IRF-7 in pDCs, 6 it was explored that whether lack of IRF-7 translocation observed is associated with PI3K/akt suppression in the presence of HIV. 2 × 10 4 pDCs/100 μL in pDC culture medium were exposed to HIV-1 with appropriate controls, such as Resiquimod (positive control) and unexposed pDCs (negative control), to study the effect on AKT phosphorylation after 20 min of incubation by flow cytometry. Histograms for pDCs showing p-AKT for each exposure were plotted, and shift of the curve toward right and increase in mean fluorescence intensity (MFI), compared with negative control, were considered as positive responses. Resiquimod induced a right shift in the peak of the histogram (green line) and an increase in MFI (2.35) compared with the negative control (blue line, MFI: 1.75) (Fig. 1D, E). On the contrary, exposure to HIV neither induced a shift of the peak (red color) nor showed an increase in MFI (1.54) when compared with the negative control (blue line, MFI: 1.75) (Fig. 1D, E). This indicates that HIV-1 may be involved in the inhibition of AKT phosphorylation or possibly may have failed to induce akt phosphorylation due to low viral dose and thus associated with the absence of IRF-7 translocation in the presence of HIV.
HIV-1 exposure causes reduction in AKT phosphorylation and lack of IRF-7 translocation induced by TLR-7 agonist
To resolve whether the inhibition of AKT phosphorylation and IRF-7 translocation associated with HIV-1 exposure was due to active inhibition or due to usage of low viral dose, pDCs (2 × 10 4 /100 μL of pDC culture medium) from four healthy donors were exposed to HIV-1 and Resiquimod simultaneously or to Resiquimod after pre-exposure to HIV for 2 h, along with exposures such as HIV-1 alone and assay control like Resiquimod alone and unexposed pDCs as media control. Phosphorylation of AKT was detected by flow cytometry after 20 min of exposure. The results demonstrated no phosphorylation of AKT after HIV exposure (Fig. 2A, B, red line, MFI: 87.9), whereas robust AKT phosphorylation (Fig. 2A, B, green line, MFI: 202) was observed with Resiquimod. However, HIV exposure in the presence of Resiquimod showed reduction in AKT phosphorylation as shown by lesser shift of histogram (Fig. 2A, purple and orange lines) to right and decreased MFI (138 and 95.9, Fig. 2A, B) compared with Resiquimod exposure alone (Fig. 2A, B, green line, MFI: 202). Furthermore, the graph shows that pre-exposure to HIV followed by Resiquimod caused enhanced suppression of AKT phosphorylation (orange line, MFI: 95.9) than simultaneous exposure to HIV and Resiquimod (purple line, MFI: 138). These results suggest that the inhibition of AKT phosphorylation observed after HIV exposure was attributable to active participation of HIV.

To observe the action of HIV on IRF-7 translocation in the presence of TLR-7 agonist, pDCs (2 × 10 4 /100 μL of pDC culture medium) were exposed to Resiquimod or HIV-1 alone or Resiquimod after pre-exposure to HIV or left unexposed. Confocal microscopic images after 3 h of exposure showed no translocation of IRF-7 into the nucleus after exposure to media control (Fig. 2C-a, D; mean fluorescence of 59787 AU in the cytoplasm and 8436 AU in the nuclear compartment) or HIV-1 (Fig. 2C-b, D; mean fluorescence of 70807 AU in the cytoplasm and 9309 AU in the nuclear compartment), whereas Resiquimod exposure caused IRF-7 to translocate into the nucleus (Fig. 2C-d, D; mean fluorescence of 4755 AU in the cytoplasm and 77078 AU in the nuclear compartment), as also seen in earlier experiment. But pre-exposure of pDCs to HIV-1 for 2 h before exposure to Resiquimod led to the inhibition of translocation of IRF-7 causing cytoplasmic arrest of this protein (Fig. 2C-c, D; mean fluorescence of 77328 AU in the cytoplasm and 9439 AU in the nuclear compartment). This underlines the active contribution of HIV-1 in the inhibition of IRF-7 translocation, which may be operational through PI3K/akt suppression by HIV-1.
HIV-1-associated inhibition of AKT phosphorylation leads to IFN-α suppression
Pure pDCs (4 × 10 4 cells/200 μL of pDC culture medium) from four healthy donors were exposed to HIV-1 alone or with HIV-1 and Resiquimod simultaneously or Resiquimod after preincubation with HIV-1 for 2 h. After 20 min, half of the cells were analyzed for p-AKT by flow cytometry and remaining half were cultured further for 8 h for IFN-α quantitation in the supernatants. Representative histogram for AKT phosphorylation and graph for average values of IFN-α from four healthy donors depict that pDC culture in which HIV-1 exposure caused the inhibition of AKT phosphorylation (Fig. 3A, red line, MFI: 1.79) also showed the absence of IFN-α production (Fig. 3B, null IFN-α), whereas exposure to Resiquimod alone caused AKT phosphorylation (Fig. 3A, green line, MFI 3.76) with downstream robust secretion of IFN-α (Fig. 3B, IFN-α: 5,877 pg/mL). pDCs showed more pronounced suppression of Resiquimod-induced phosphorylation of AKT when they were preincubated for 2 h with HIV-1 (Fig. 3A, orange line, MFI: 2.31) than simultaneous exposure to HIV-1 and Resiquimod (Fig. 3A, purple line, MFI: 2.96), and similar phenomenon was observed for IFN-α as well (Fig. 3B, TLR-7 agonist + HIV: 2,821 pg/mL, Resiquimod after preincubation with HIV: 538 pg/mL). When average values of MFIs for each exposure and IFN-α secreted by four donors were compared, the inhibitory effect on p-AKT was found to be positively associated with suppressed IFN-α production (Fig. 3B). The results suggest that the inhibition of TLR-7 agonist-induced phosphorylation of AKT by HIV may have lead to decrease in IFN-α in pDCs.
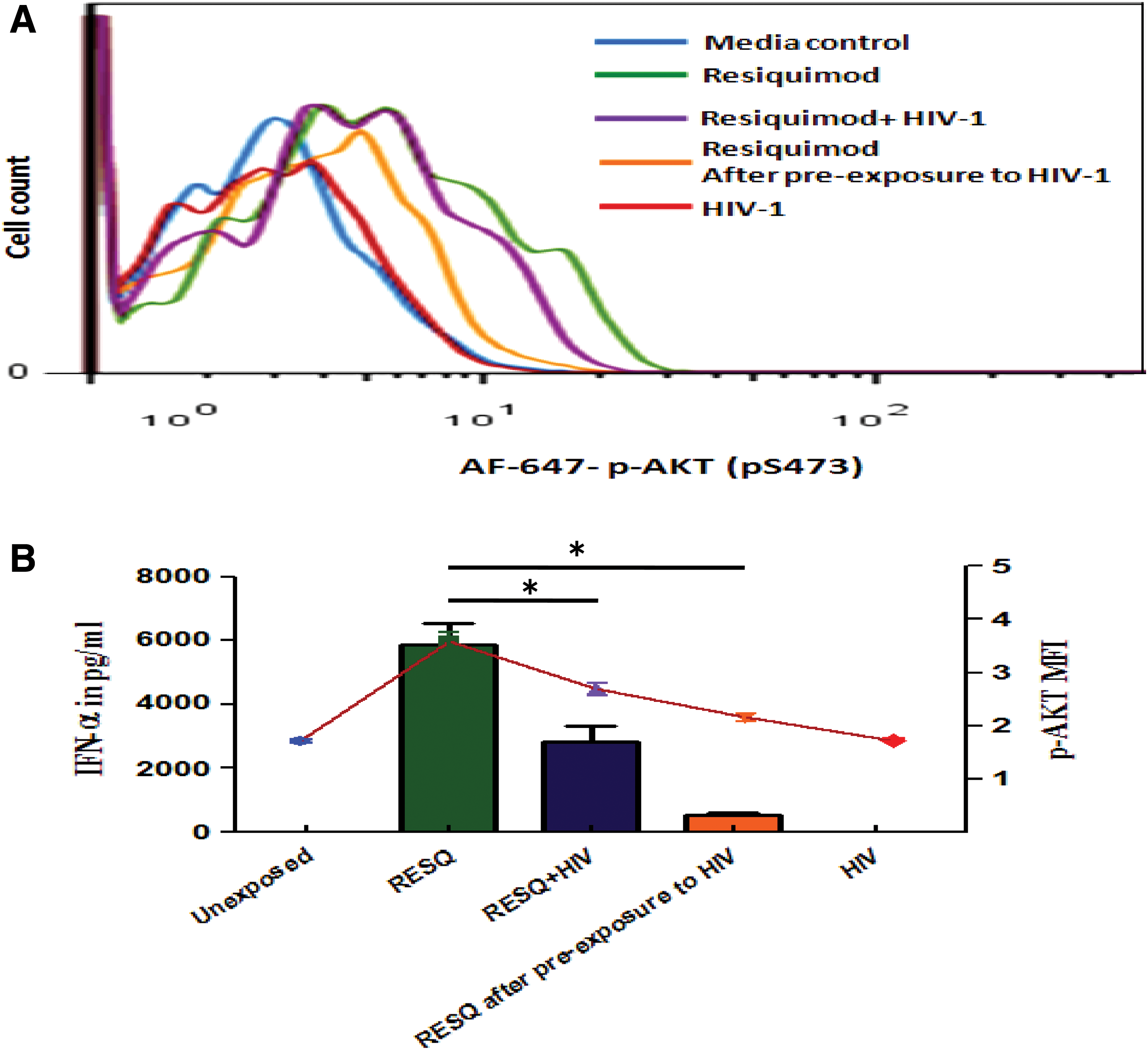
Graphs from flow cytometry and ELISA assays depicting the association of HIV-1-induced p-AKT inhibition and IFN-α suppression: pure pDCs were exposed to different conditions. After 20 min, half the number of pDCs were fixed, permeabilized, and stained; the remaining pDCs were incubated for 8 h and IFN-α in supernatants was quantified by ELISA.
HIV exposure to pDCs causes PI3K/akt inhibition leading to the inhibition of IRF-7 translocation and IFN-α suppression
To observe the effect of HIV on serial events of TLR-7 activation pathway, 6 × 10 4 pDCs were exposed to Resiquimod or HIV-1 or left unexposed in 300 μL of pDC culture medium in CO2 incubator. After 20 min, one-third of the cells from each well were analyzed for p-AKT by flow cytometry, whereas after 3 h of exposure, half of the cells from remaining population were stained for IRF-7 for confocal microscopy and supernatants from remaining pDCs were analyzed for IFN-α levels by ELISA after 8 h. Results showed that, in HIV-exposed pDCs, AKT phosphorylation was absent (Fig. 4A, red line), which led to the inhibition of IRF-7 translocation (Fig. 4B-b, C; mean fluorescence of 55254 AU in the cytoplasm and 13315 AU in the nuclear compartment) and ultimately resulted in IFN-α suppression (Fig. 4D). Media control (unexposed pDCs) showed no IFN-α secretion (Fig. 4D) with the absence of p-AKT (Fig. 4A) and translocation of IRF-7 (Fig. 4B-a, C; mean fluorescence of 61423 AU in the cytoplasm and 25445 AU in the nuclear compartment) as against robust IFN-α secretion in association (Fig. 4D) with AKT phosphorylation (Fig. 4A) and IRF-7 translocation by positive control (Resiquimod) (Fig 4B-c, C; mean fluorescence of 5445 AU in the cytoplasm and 83514 AU in the nuclear compartment). These results support our hypothesis that HIV may be using PI3K/akt pathway inhibition strategy to cause inhibition of IRF-7 translocation to suppress IFN-α production.
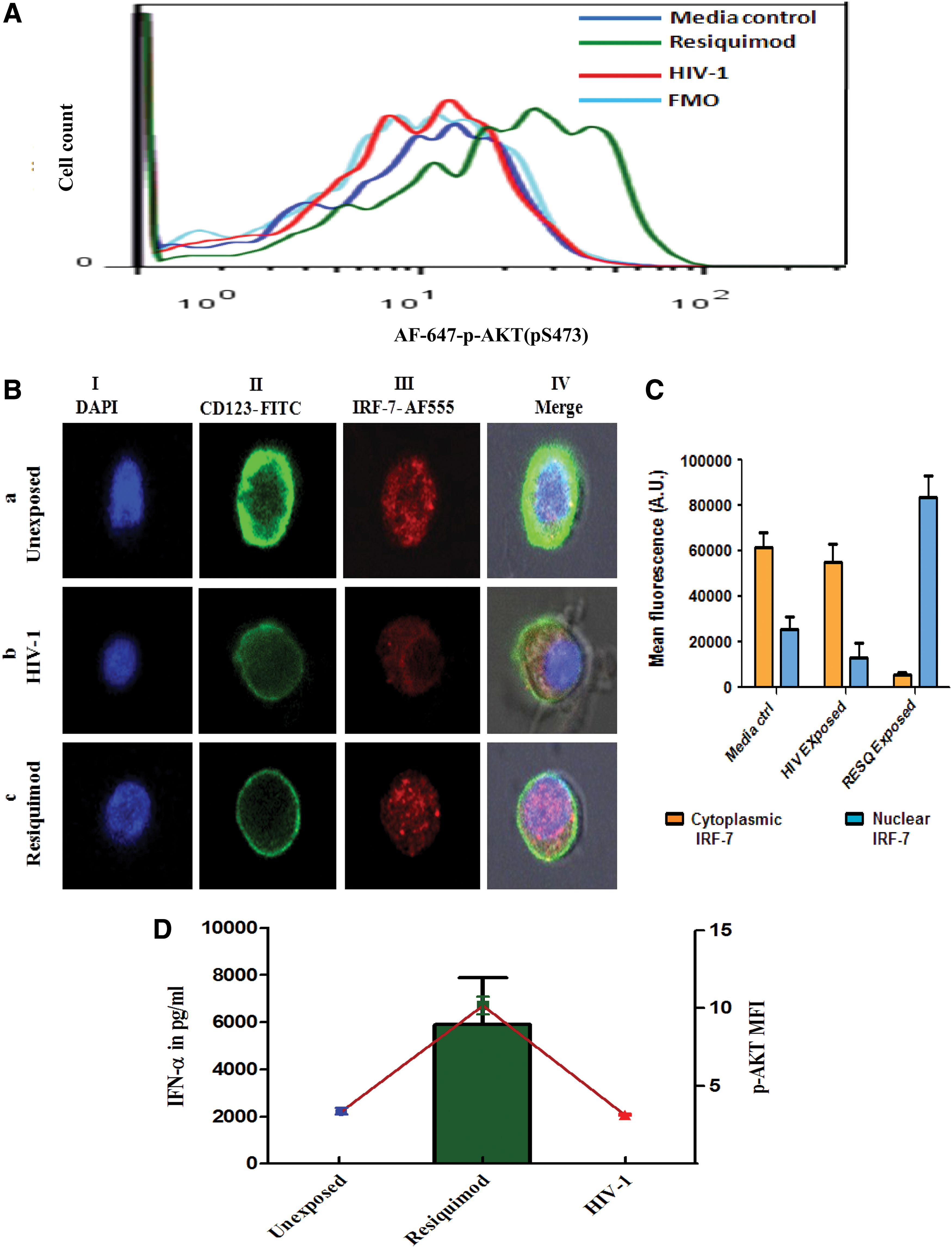
Simultaneous detection of IRF-7 translocation, p-AKT, and IFN-α production in cultured pDCs: pure pDCs were analyzed for p-AKT, IRF-7 translocation, and IFN-α secretion at specified time points after exposure to TLR-7 agonist or HIV-1 or with no exposure in the same pDC culture.
Discussion
Engagement of TLR-7 by microbial ligands induces a cascade of events that lead to the activation of IRF-7, the transcription factor for IFN-α, and subsequent expression and release of IFN-α, a cytokine critical for innate immune responses. 8 IRF-7 is the key protein, which upon getting signals from upstream signaling molecules gets phosphorylated and translocates from cytoplasm to nucleus to activate residing IFN-α genes. AKT phosphorylation of PI3K/akt pathway is crucial for the translocation of IRF-7 into the nucleus in pDCs, as shown previously. 6 We explored the influence exerted by HIV-1 at key levels of TLR-7 signaling, activation of PI3K/akt pathway by AKT phosphorylation, translocation of IRF-7 into the nucleus, and release of IFN-α. Our findings suggest that the suppression of IFN-α production caused due to exposure of pDCs to HIV-1 is attributable to the inhibition of AKT phosphorylation, which leads to the suppression of translocation of IRF-7 into the pDC nucleus, a key step for IFN-α production.
A number of in vitro studies have reported the production of IFN-α in response to exposure of pDCs to HIV-1. 9 –11 However, these studies have used virus copies at higher levels than seen in the acute HIV infection. 12 –16 Our recent studies have provided evidence that in vivo doses of HIV-1 matching with viral copies seen in acute HIV infection suppress the production of IFN-α from pDCs. 7 Hence, in this study, we have exposed pDCs to HIV virus copy number similar to that seen in the acute primary HIV infection to explore the effects of HIV exposure on cell signaling events crucial for IFN-α gene induction, i.e., IRF-7 translocation and AKT phosphorylation.
To check the functionality of TLR-7 pathway in the presence of HIV-1, we first evaluated the effect of HIV-1 on IRF-7 translocation in pDCs. IRF-7 was found to be located in the cytoplasm even after 3 h of exposure of pDCs to HIV-1, in contrast with the translocation of IRF-7 into the nucleus in response to TLR-7 agonist, thus indicating that HIV-1 may be involved in the inhibition of translocation of IRF-7. Failure of HIV to induce TLR-7 cell signaling pathway may be attributable to low copy number used to induce pDCs in our study or to an active inhibitory role of HIV. This possibility was resolved in our subsequent experiments, where we found that pre-exposure of pDCs to HIV almost completely inhibited Resiquimod-induced IRF-7 translocation. This shows active participation of HIV in the inhibition of IRF-7 translocation, rather than mere failure of virus to activate TLR-7 cell signaling cascade.
It has been reported that AKT phosphorylation in PI3K/akt pathway is required for the translocation of IRF-7 into the pDC nucleus. 6 In present study, AKT phosphorylation was found to be inhibited upon exposure to HIV-1, and this inhibition was seen even when pDCs were exposed to TLR-7 agonist in the presence of HIV-1. Thus, the results again support the active role of HIV in the inhibition of PI3K/akt pathway. The pDCs, which were preincubated with HIV-1, demonstrated higher inhibition of AKT phosphorylation than those incubated simultaneously with HIV-1 and TLR-7 agonist. Furthermore, we tested whether this HIV-1-linked inhibition of AKT phosphorylation leads to IFN-α abrogation or not. It was observed that pDCs, which showed HIV-1-associated inhibition of AKT phosphorylation, also showed complete suppression of IFN-α secretion. Additionally, abrogation of TLR-7 agonist-induced p-AKT by HIV-1 resulted in IFN-α inhibition, proportional to p-AKT suppression, i.e., preincubation with HIV-1 for 2 h caused higher inhibition of AKT phosphorylation as well as IFN-α production than simultaneous incubation of HIV-1 and TLR-7 agonist. These results are consistent with earlier findings from our study where higher inhibition of TLR-7 agonist-induced IFN-α production was seen in pDCs preincubated with HIV-1 than those simultaneously incubated with HIV-1 and TLR-7 agonist. 7 All these findings indicate that IFN-α suppression observed in the presence of HIV exposure may be attributable to HIV-induced inhibition of AKT phosphorylation of PI3K/akt pathway. The findings additionally support the fact that HIV-1-exposed pDCs cannot be stimulated again with TLR-7 agonist to produce IFN-α, which is contradictory to the study of O'Brien et al. showing IFN-α production by HIV-1-exposed pDCs upon re-exposure to other viruses. 17
Our findings showed that the presence of HIV was associated with the inhibition of IRF-7 translocation as well as inhibition of PI3K/akt pathway leading to IFN-α abrogation. But an evidence showing abrogation of serial events of TLR-7 pathway, phosphorylation of AKT, IRF-7 translocation, and IFN-α secretion was absent. Hence, to fill this gap, HIV-1-exposed pDCs in the presence of appropriate controls were assessed initially for AKT phosphorylation, subsequently for IRF-7 translocation, and finally for IFN-α production from same pDC culture. Results showed that HIV-exposed pDCs display inhibition of AKT phosphorylation, which led to abrogation of IRF-7 translocation resulting into IFN-α suppression. This strongly indicates that HIV exposure takes PI3K/akt inhibition pathway to block IRF-7 translocation causing IFN-α abrogation. This is also supported by the fact that there was quantitative association between the suppression of AKT phosphorylation and reduced IFN-α production by pDCs upon exposure to HIV. Hence, the strategy used by HIV to inhibit AKT phosphorylation to suppress IRF-7 translocation and IFN-α production in pDCs may be responsible for IFN-α impairment observed in recently HIV-1-infected individuals. However, the causal relationship of HIV exposure and inhibition of AKT phosphorylation and IRF-7 inhibition is yet to be conclusively demonstrated. The inhibition of AKT phosphorylation in the presence of HIV has been suggested in earlier study by Huang et al., 18 where TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis requiring AKT inhibition was compared between HIV-infected and noninfected monocyte-derived macrophages (MDM). The results showed that HIV-infected MDM showed inhibition of AKT phosphorylation and were more susceptible to TRAIL-induced apoptosis than uninfected MDM, suggesting the role of HIV in AKT inhibition in infected MDM. 18
While we used whole HIV virus particles in our studies, it will be interesting to know whether one or more specific viral proteins are responsible for the suppression of IFN-α production. HIV-1 proteins, Vpr, vif, and Tat were found to inhibit the action of IRF-1 and IRF-3 proteins by targeting them for proteosomal degradation, 19,20 whereas HIV-2 Vpx protein also inhibited the action of IRF-5 to cause the suppression of type-I interferons. 21 Considering the effect of these HIV proteins on IRFs, the role of these and other HIV-1 proteins in alteration of p-AKT and so IRF-7 needs to be explored for better understanding of IFN-α inhibition in pDCs. Additionally, other than blocking PI3K/akt pathway, HIV-1 may use other strategies for blocking IFN-α production, such as BDCA-2 ligation to HIV-1 envelope gp120, which results in IFN-α suppression in pDCs. 7,22,23 The reduced IFN-α production observed in pDCs induced by Resiquimod after pre-exposure to HIV-1 in our experiments may reflect abrogated TLR-7 recycling due to HIV exposure, and hence, less of it would be available to detect and respond to subsequent Resiquimod challenge. Further studies for exploring such possibilities are needed. Searching for direct evidence for HIV effects on cell signaling cascades as well cellular proteins will help in understanding IFN-α inhibition and pDC-HIV interaction in recent HIV-1 infection.
The results obtained here can be related to in vivo situation occurring after initial exposure of HIV-1, where pDCs get recruited at the site of infection, but HIV-1 through its action on AKT and IRF-7 suppresses IFN-α production, facilitating spread and successful establishment of HIV-1 infection. The functional loss of pDCs caused by HIV-1 cannot be reverted back even after successful antiretroviral treatment or by stimulation by TLR-7 agonist, as shown here and earlier from our laboratory. 5 But the long-term nonprogressors (LTNPs) show minimal functional defects in pDCs and also do not show decline in pDC numbers in circulation. 5 It is possible that LTNPs have managed to preserve pDC functionality during recent HIV infection, aiding in achievement of better virus control. The condition of decline in pDC numbers and functions cannot be improved even in the presence of successful chemotherapy in HIV-infected patients. Hence, the preservation of pDC numbers and functions during the early HIV infection may be the key for better virus control. As this study shows the role of HIV-1 in compromising pDC functions, understanding full repertoire of mechanisms used by HIV to compromise the pDC numbers and functions is likely to provide new targets for immunological interventions.
Footnotes
Acknowledgments
We thank all healthy donors for donating their blood and Sahyadri Blood Bank, Deccan Gymkhana, Pune, for providing buffy coats during this study. The facilities and guidance provided by Dr. Patole, Mrs. Ashwini, Mrs. Trupti, and Dr. Wani of the National Centre for Cell Sciences, Pune, are acknowledged. We also express our gratitude toward the Indian Council of Medical Research (ICMR), Delhi, for financial support to A.S.D.
Authors' Contributions
A.S.D. and R.S.P. have conceptualized the study and wrote the article. All laboratory experiments were performed, analyzed, and interpreted by A.S.D., with the aid of R.S.P. and M.R.T. All authors have substantial contribution in execution of laboratory work, critical analysis of results, and drawing conclusions. R.S.P. and M.R.T. have approved the final version of the article and agreed to be responsible for accuracy and integrity of the work carried out in this study.
Author Disclosure Statement
These data have not been presented or published elsewhere. The authors have no competing interests or disclosures.