
Abstract
The MWRI-01 study characterized the safety, acceptability, pharmacokinetic (PK), and pharmacodynamic (PD) profile of rilpivirine (RPV) long acting (LA) in a model of preexposure prophylaxis (PrEP). Prospective, open-label Phase 1 study. The safety and acceptability of three repeated doses of RPV LA were monitored. Blood, tissue (rectal, cervical, and vaginal), and biological fluids (vaginal and endocervical) were collected at baseline and at 1- to 2-month intervals throughout the study for PK and PD assessment. Eight women and four men received three intramuscular doses of 1,200 mg of RPV LA given 8 weeks apart. There were a total of 195 adverse events (AEs) reported, of which 138 (70.8%) were Grade 1 and 55 (28.2%) were Grade 2. The most common AE was injection site pain. Geometric mean (90% confidence interval) plasma RPV concentrations at 56 days after the first and third doses were 39 (33–45) ng/mL (female)/29 (17–40) ng/mL (male) and 59 (45–62) ng/mL (female)/40 (30–51) ng/mL (male), respectively. Exposure to RPV LA was associated with significant inhibition of HIV-1BaL viral replication in the ex vivo rectal explant model (p < .0001) that persisted for up to 4 months after the third dose of RPV LA. In contrast, no viral suppression was seen in cervicovaginal tissue. Multiple dose administration of RPV LA was safe and well tolerated, and was associated with prolonged suppression of viral replication in rectal explant tissue.
Introduction
Oral and topical antiretroviral preexposure prophylaxis (PrEP) is safe and effective in preventing HIV-1 infection in a broad range of at-risk populations. 1 –4 However, PrEP efficacy is significantly diminished when recipients have suboptimal adherence. 5 Consequently, long-acting (LA) injectable PrEP is currently being developed to provide an alternative for individuals who require PrEP, but are unable or unwilling to be adherent to oral or topical formulations. 6,7 Rilpivirine (RPV), a non-nucleoside reverse transcriptase inhibitor (NNRTI), has previously been investigated in Phase 1 and 2A studies. 8,9 Cabotegravir LA, an integrase inhibitor, has advanced to Phase 2B/3 safety and evaluation (HPTN 083. NCT02720094), and EFdA, a nucleoside reverse transcriptase inhibitor, 10 has shown efficacy in animal models of HIV infection 11 as well as early Phase 1 human studies. 12 RPV LA and cabotegravir LA are also being developed as the first dual agent LA injectable antiretroviral therapy for the treatment of chronic HIV-1 infection. 13
Oral RPV is currently licensed for the treatment of HIV-1 infection. 14 NNRTIs are attractive PrEP agents because they act early in the viral replication cycle, are potent antiretroviral agents, and, unlike tenofovir (TFV), do not require cellular metabolism to be active against HIV. Snyder et al. showed that immunodeficient mice reconstituted with human lymphoid tissue and treated with RPV LA were protected from infection when challenged vaginally with HIV-1CH040. 15 We have previously reported on the safety and pharmacokinetics (PK)/pharmacodynamics (PD) of single-dose RPV LA, 9 and in this article, we have expanded these observations with data on multiple injections of RPV LA. In addition, we also characterized the efficacy of RPV against both Clade B and Clade C HIV-1 infection in the colorectal and cervicovaginal ex vivo challenge model.
Methods
Study design
The MWRI-01 study was a Phase 1, single-center, prospective, open-label exploratory study of RPV LA conducted in healthy HIV-1-seronegative men and women. The study was originally designed in multiple phases. The first phase of the study explored single doses of RPV LA (600 and 1,200 mg) and was conducted between November 2013 and August 2014.
9
The second phase of the study is described here and explored three injections of 1,200 mg of RPV LA. Screening and enrollment for this phase of the study began in November 2014 and follow-up was completed in January 2016. The study was registered at
Setting and participants
Participant enrollment occurred at the Clinical and Translational Research Center at the Magee-Womens Hospital, Pittsburgh, PA. Participants were initially eligible for inclusion if they were 18 through 45 years of age, had a body mass index (BMI) between 18 and 35 kg/m2, were HIV-1 seronegative, and were willing to use contraception. Females were not pregnant or breastfeeding, had a regular menstrual cycle, and had satisfactory cervical cytology. Males had to be willing to abstain from sperm donation for the duration of the study. Major inclusion and exclusion criteria were the same as in the single-dose phase of the study and summarized in our previous publication. 9
Study product
RPV LA (300 mg/mL) was administered as two 2 mL (a total of 1,200 mg) intramuscular (IM) gluteal injections. The two injections were given ∼1 h apart. The injections were given without any oral RPV lead-in in any participant. RPV LA was supplied in vials as a sterile aqueous nanosuspension formulation (G001) at a concentration of 300 mg/mL (Janssen Pharmaceutica NV, Beerse, Belgium). Before use, the RPV LA was stored in a temperature-controlled environment (2°C–8°C).
Study procedures
There were a total of sixteen study visits. After obtaining informed consent, all participants were screened [Visit (V) 1] with a medical history, a physical examination, including rectal and vaginal examination, and rectal swabs for Chlamydia trachomatis (CT)/Neisseria gonorrhoeae (GC) nucleic acid amplification testing (NAAT). Urine was also collected for CT/GC NAAT and for pregnancy testing in female participants (pregnancy testing was repeated at all subsequent clinical visits). Blood was collected for safety labs (complete blood count, prothrombin time, urea nitrogen, creatinine, ALT, and AST) and serology [syphilis, HIV-1, HBsAg, HCV, and herpes simplex virus (HSV) 1 and 2]. A 12-lead ECG was recorded in triplicate.
Participants who met the inclusion/exclusion criteria during the screening visit were enrolled into the study. The enrollment/baseline visit (V2) occurred within 28 days of screening. At V2, a web-based behavioral questionnaire was administered and a focused physical examination was performed. Rectal swabs, urine, and blood samples were collected for CT/GC NAAT, pregnancy testing, and HIV-1 serology. Vaginal and endocervical fluid (EF) samples were collected using Merocel sponges (Medtronic, Mystic, CT) for preexposure PK and ex vivo PD assays.
Participants then received a 125-mL normal saline pH 7.4 enema. Flexible sigmoidoscopy was performed with collection of 10 rectal biopsies at ∼15 cm from the anal verge as previously described. 16 –18 Biopsies were collected using large-cup endoscopic biopsy forceps with an outside diameter of 3.3 mm (Microvasive Radial Jaw No. 1589; Boston Scientific, Marlborough, MA). Female participants had two ectocervical and three vaginal biopsies. Biopsies were used for PK and PD (ex vivo tissue challenge).
Participants then received two 600 mg IM injections of RPV LA separated by an hour. Blood and genital tract fluids were collected 24 h (V3), 1 week (V4), and 4 weeks (V5) after the dosing. The second administration of 1,200 mg RPV LA was 8 weeks later at V6 (day 56) and the third 8 weeks later at V10 (day 112). Blood was also collected 1 day after the two additional injection visits (V7 and V11), and at all additional study visits, including V8 (day 63), V9 (day 84), and V12 (day 119). Rectal, cervical, and vaginal tissue (VT) were collected at V2 (Baseline), V6 (day 56), V10 (day 112), V13 (day 140), V14 (day 168), V15 (day 196), and V16 (day 224). The rectal biopsies were optional for the female participants.
Clinical safety and laboratory assessments
Adverse events (AEs) were graded using the Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 1.0, December 2004, and Addenda 1–3 (
Behavioral evaluations and procedures
A web-based self-interview, also known as a computer-assisted self-interview (CASI), was completed by the participant at V2. In addition to demographics, this questionnaire also assessed each participant's history of HIV sexual risk behaviors and experience with safer sex practices. The CASI included questions about history of alcohol use and other psychoactive substances. Participants completed additional CASIs after the injections at V3, V7, V11, and V16. These questionnaires included structured and semi-structured questions about experiences the participant had after receiving RPV LA, including an assessment of general product acceptability. Participants were asked to rate acceptability on a scale from 1 to 10, with 1 representing less acceptable/unlikely. Participants were also asked several questions related to both the product and perceived barriers to product use.
PK analysis
RPV concentrations in all matrices [plasma, mucosal fluids (endocervical and vaginal), and tissue (rectal, ectocervical, and vaginal)] were quantified by validated high-pressure liquid chromatography–mass spectrometry using a Thermo triple quadrupole TSQ Vantage mass spectrometer (Thermo Electron Corporation, Hemel Hempstead, United Kingdom), operating in the positive ionization mode (selected reaction monitoring). The tissue processing pathway for samples from the MWRI-01 has previously been described elsewhere with full methodological validation of this process. 19
RPV concentrations in all matrices were expressed as ng/mL. Tissue homogenate and vaginal/EF samples were initially quantified using an ng/sample calibration curve and then converted to ng/mL by adjusting for the recorded tissue and fluid volumes. Concentrations that were below the assay lower limit of quantification (LLOQ; 0.5 ng/mL) were expressed as 1/2 LLOQ values.
Ex vivo tissue biopsy HIV-1 challenge assay
At baseline (V2) and postproduct exposure time points (V6, V10, and V13–V16), rectal, cervical, and vaginal biopsies were collected in 20 mL RPMI and transported to the laboratory for ex vivo infection within 30 min using a common viral stock of the Clade B virus HIV-1BaL (105 TCID50 for rectal and 5 × 104 TCID50 for cervicovaginal tissue), as previously described. 20 –23 Rectal explants were also challenged in a similar manner with 1.79 × 106 TCID50 of virus from an anonymized subtype C HIV-1-infected treatment-naive individual from South Africa. Viral RNA was extracted from the sample, amino acids 1–560 of reverse transcriptase were amplified, and the full-length reverse transcriptase gene was bulk cloned into an HIV-1LAI viral vector that had undergone silent mutations to introduce two cloning restriction sites. Virus was produced by Lipofectamine 2000 transfection of 293T cells. A full sequence was done on the original plasma virus to verify wild-type genotype, and the intermediate plasmid pretransfection and laboratory-generated virus were also genotyped to make sure the sequences were identical and clustered with the virus in the original plasma sample. 24
Ex vivo culture supernatants for p24 quantification were collected every 3–4 days over the 14-day culture period (days 4, 7, 11, and 14). Results were adjusted for initial biopsy weight, for rectal biopsies averaged across quadruplicate assays, after statistical outliers were excluded at the 95% confidence level for the Dixon outlier test and reported as day 14 cumulative p24 (p24 HIV antigen enzyme-linked immunosorbent assay; Alliance; Perkin-Elmer Life Sciences, Inc., Boston, MA) where the assay's LLOQ was 10 pg/mL. Nondetectable cumulative p24 measures on day 14 were converted to 1/2 LLOQ (i.e., 5 pg/mL) before log transformation.
Statistical analyses
Given the small sample size in this Phase 1 study, there was insufficient power to compare any AEs other than those occurring at high frequency. The primary safety endpoint in the study was the proportion of participants with a Grade 2 or higher AE. Each participant contributed once (i.e., highest severity AE for each participant) for the calculation of event rates. Acceptability of the study product was evaluated by CASIs at several time points after administration. Participants completed those CASIs at the 24-h postdose visit (V3), 7-day postdose visit (V4), and final follow-up visit (V12).
Plasma and compartmental PK endpoints included the concentrations measured at monthly intervals (C28d, C56d, C84d, and C112d postdose), area under the concentration-time curve over 2 months after each injection (AUC56d postdose), and the AUC to 4 months (AUC112d) after the third and final injection, the maximum concentration (Cmax), the time to maximum concentration (Tmax), and the terminal t1/2 following the third dose of RPV. For male participants, this included plasma and rectal tissue (RT). For female participants, this included values from plasma, endocervical and vaginal secretions, and ectocervical and VT. RT PK data were also available from those female participants who elected to have rectal samples collected.
PK parameters were calculated using noncompartmental analysis (WinNonlin Phoenix, version 6.1; Pharsight, Mountain View, CA). Measures of drug exposure and ratios of compartmental-to-systemic drug [(RPV)COMP/(RPV)Plasma] over 224 days were analyzed. Data were expressed as geometric means (GM) with 90% confidence intervals (CI). Within-participant changes in the assessed PK parameters following multiple injections were evaluated by calculating geometric mean ratios (GMRs) and associated 90% CI. The 90% CI were determined using logarithms (log10) of the individual GM values and the calculated values were expressed as linear values. The within-subject changes in PK parameters were considered to be statistically significant when the 90% CI of GMRs did not cross the value 1.
Changes in culture supernatant log10 HIV-1 p24 antigen levels across visits were compared to baseline levels using repeated measures analysis of variance (ANOVA), where the p24 antigen levels were the cumulative p24 measures up to day 14 of the ex vivo assay. Multiple sites (cervical, vaginal, and rectal) were studied, using HIV-1BaL. In addition, rectal biopsies were also challenged with a Clade C virus (G147-1).
Detectable PK drug levels and cumulative p24 antigen levels for measures taken from the same participants/visits were correlated using a linear mixed-effect model, with subject random factor, to test for a significant slope estimate for each PK:PD pair, where a negative slope would indicate potential PD activity. Statistically significant linear PK:PD relationships were further tested using a three-parameter nonlinear, logistic PK-PD model [Eq. (1)]
25
.
where E is the % virus control, EMax are the lower-upper plateaus of the curve, and c is the drug concentration.
The parameters for lower, upper, and EC50 were estimated by PROC NLIN (SAS/STAT® Software) and the % virus control was calculated using visit 2 data as each participant's virus control baseline [Eq. (2)] so that if baseline p24 = 100 pg/mL and observed p24 was 50 pg/mL, then that would be a result of 50% virus control. Percentage of virus control results found to be <0% and >100% were imputed as 0% and 100% virus control, respectively, as virus (p24) above baseline levels were considered to clearly show active growth (>100%), while virus <1/100th of the virus growth baseline (<0%) was considered to show no virus growth, no matter how high or low the actual p24 amount was, respectively, compared to baseline levels.
where p24 (pg/mL) baseline was measured from samples collected at visit 2.
Results
Participants
A total of 18 participants were screened and 12 were enrolled in the study. Participants were mostly white (9/12) with a mean age of 28.9 [standard deviation (SD) = 7.9] years and a mean BMI of 24.5 (SD = 3.7) (Table 1). All 12 of the participants (8 females and 4 males) received three 1,200 mg doses of RPV LA. Eleven of the 12 participants completed all 16 visits. One male participant left the study at V14 to attend college (Fig. 1).
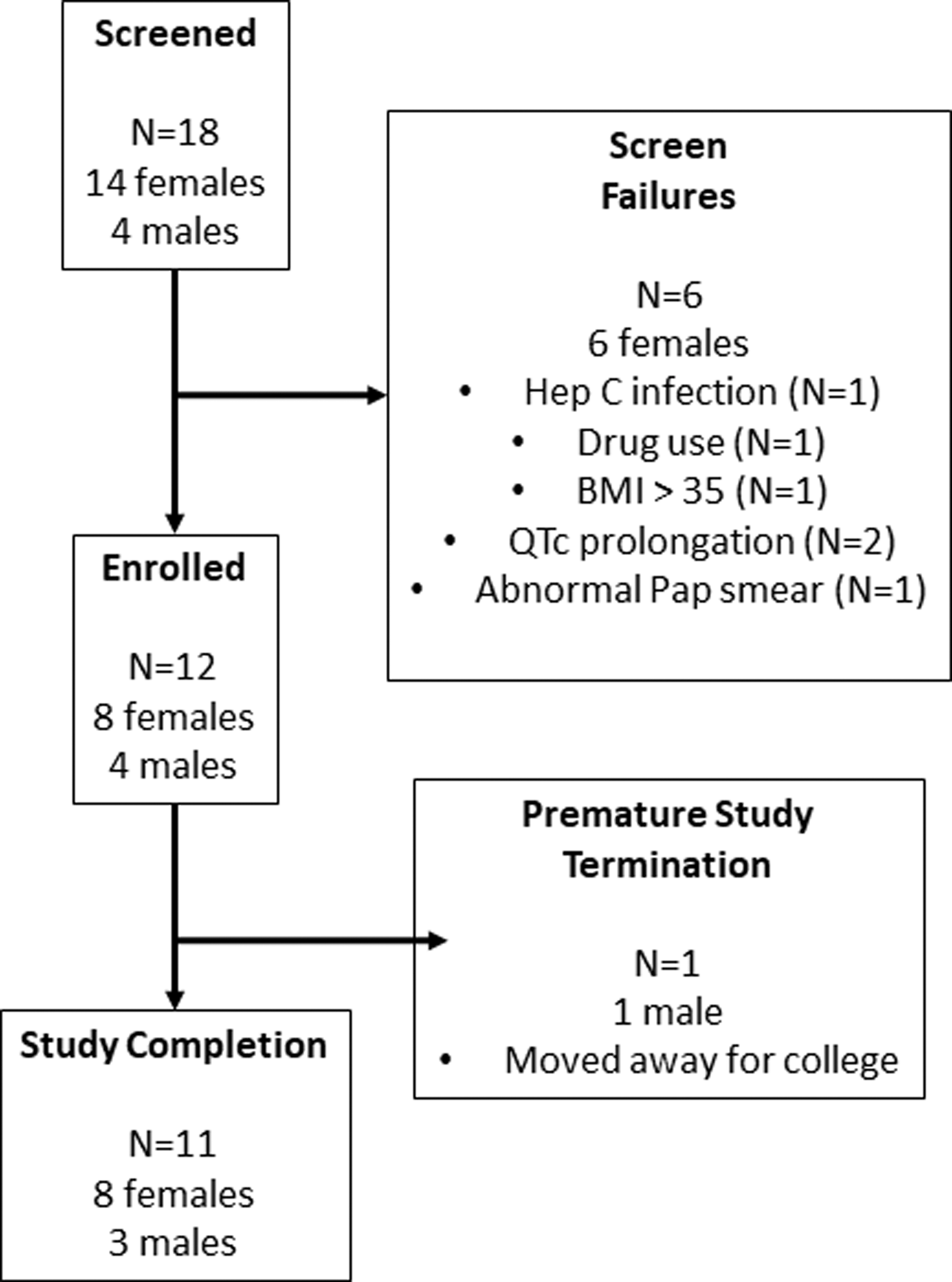
Consort diagram of MWRI-01 multidose participants.
Demographic and Clinical Characteristics, Overall and by Study Arm and Sex
BMI, body mass index; SD, standard deviation.
Safety
There were a total of 195 AEs reported, of which 138 (70.8%) were Grade 1, 55 (28.2%) were Grade 2, and 2 (1.0%) were Grade 3. Both Grade 3 AEs (a finger laceration with secondary infection) were deemed not related to study product.
Injection site reaction (ISR) was the most frequent AE and was reported in all participants (Table 2). The most commonly reported ISR was pain [Grade 1; 2/12 (16.7%) and Grade 2; 10/12 (83.3%)]. The average length of ISR AEs (±SD) was 3.31 (±2.39) days.
Injection Site Reactions
Electrocardiograms were performed throughout the study. Two of the 12 participants each had one episode where the QTc interval was above the upper limit of normal defined by the DAIDS toxicity table (450 ms). These events (452 and 454 ms) occurred at 56 and 112 days, respectively, after the third dose of RPV LA.
Behavioral evaluation and product acceptability
Eleven of the 12 participants completed Product Acceptability Questionnaire at V14 during the 4-month follow-up (3 men and 8 women; 1 male participant did not complete this instrument). In general, participants were somewhat neutral about using the product, assuming it was found to be very good at protecting people from HIV (mean = 5.27, with 1 = extremely unlikely and 10 = extremely likely). As the per injection cost of the product increased, participant enthusiasm decreased; at $25, the mean score was 4.18, at $50, the mean score was 2.30, and finally, at $100, the mean score was 1.50.
Overall, participants reported moderate fear of injection (mean = 5.64, with 1 = extremely and 10 = not at all fearful/nervous) and experience of painful injection (mean = 5.27, with 1 = extremely painful and 10 = not at all painful). Of the suggested barriers to uptake, participants were most concerned with cost (mean = 8.5), possible medication side effects (mean = 8.09), and fear that medications could be harmful to their health (mean = 6.91). Less of a concern for participants were the ability to protect oneself without their partner knowing (mean = 4.0), fear of needles (mean = 3.91), and stigma associated with using an HIV medication (mean = 3.09).
PK analysis
PK profiles (GM; 90% CI) for RPV in plasma and mucosal sites [vaginal fluid (VF), EF, VT, ectocervical tissue (ET), and RT] of females and males (RT only) over 224 days after three injections of RPV LA at 2-month intervals were determined (Fig. 2). Plasma and tissue PK parameters and associated GMRs are presented in Table 3. VF and EF PK data are provided in Supplementary Table S1. The target concentration for HIV prevention is unknown, but for comparison, we utilized the RPV in vitro EC90 for wild-type HIV, corrected for protein binding to yield a putative target concentration of 12.2 ng/mL. 26

Pharmacokinetic profile. RPV concentrations in male and female study participants over 224 days after three injections of RPV LA every 2 months.
Rilpivirine plasma and Tissue Pharmacokinetic Parameters and Geometric Mean Ratios
RPV PK parameters following second and third dose compared with that of the first dose (single dose).
RPV PK parameters following third dose compared with that of the previous dose (second dose).
AUC56d, area under the concentration-time curve over 2 months postdose; C56d, concentration 2 months postdose; CI, confidence interval; Cmax, maximum concentration; ET; ectocervical tissue; F, females; GM, geometric mean; GMR, geometric mean ratio; m, males; PK, pharmacokinetic; RPV, rilpivirine; RT, rectal tissue; VT, vaginal tissue.
Plasma
Maximum RPV plasma concentrations (Tmax) were reached within 7 days postdose following both single (first dose) and multiple (second and third dose) injections of RPV LA. The plasma terminal t½ was ∼70 days in males and females. GM (90% CI) plasma RPV concentrations at 2 months (C56d postdose) after the first and third doses of RPV LA were 39 (33–45) ng/mL (female)/29 (17–40) ng/mL (male) and 59 (45–62) ng/mL (female)/40 (30–51) ng/mL (male), respectively.
Plasma concentrations >EC90 were achieved for all subjects, irrespective of dosing occasion or gender, at the time of the first PK sampling at 24 h postdose (days 1, 57, and 113) and at 2 months at the time of the next injection (days 56 and 112). All concentrations remained above the plasma EC90 for up to 112 days (day 224) after the third injection: GM (90% CI) plasma C112d postdose were 37 (26–47) ng/mL (female) and 27 (20–34) ng/mL (male). Furthermore, by extrapolating from the last measurable time point using the individual terminal t½, the median estimated time to the LLOQ (0.5 ng/mL) was ∼15 months after the final injection, although two subjects had plasma levels that persisted for a longer period.
Overall, there was evidence of accumulation in plasma after the second dose with an approximate 30% increase in RPV exposure (AUC56d) in females and a 25%–45% increase in plasma levels (C56d/Cmax) in males. However, the amount of accumulation, following the second and third doses, was not consistent for all evaluated PK parameters (Table 3), which is likely attributed to the study's sparse sampling schedule.
RPV plasma exposures (cumulative AUC following three doses) were, on average, 36% lower in males compared with females (p = .042). Plasma exposures (AUC56d postdose) following the first dose were equivalent to those observed in the single-dose arms (1A/1B) during the first phase of the MWRI-01 study in subjects receiving an equivalent 1,200 mg dose (females p = .208; males p = .762). Five subjects had low, detectable levels of drug in plasma before the first dose and the analysis repeated excluding those subjects (Plasma) provided a similar result (p = .71, slope = −0.13, r 2 = 0.00, n = 3).
Female genital tract
In the female genital tract, single-dose and multidose RPV exposures in VF were consistently higher than plasma and increased (35% VF and 48% EF) following the second injection (Supplementary Table S1). RPV GM genital fluid-to-plasma ratios at 2 months (C56d postdose) after the first, second, and third doses were 1.51, 2.19, and 2.17 for VF and 0.85, 1.17, and 0.92 for EF.
Tissue
Tissue biopsies (VT, ET, and RT) were taken at two monthly intervals (days 56 and 112) just before the next dosing occasion and at monthly intervals (days 140, 168, 196, and 224) following the third injection (Fig. 2C, D). All eight female participants opted to undergo RT sampling.
RPV GM tissue-to-plasma ratios at 2 months (C56d postdose) after the first, second, and third doses were 0.56, 0.78, and 0.67 for VT and 0.73, 0.83, and 0.74 for ET. For RT, ratios were 1.17, 1.30, and 1.12 (females) and 1.23, 1.46, and 1.02 (males). In females, RT levels, expressed as ng/mL, were on average 1.5- to 2-fold higher than corresponding RPV tissue concentrations in the female genital tract (VT p < .02; ET p > .109).
Significant RPV accumulation was observed in VT (68% increase), ET (37% increase), and RT (49% increase; males only) after the second dose (Fig. 2C, D).
Pharmacodynamics
Ex vivo HIV-1 infection (log10 p24 pg/mg) in RT, ET, and VT was compared between baseline (V2) and days 56–224 after IM injection of RPV LA (Fig. 3) by mixed-model ANOVA. Pairwise comparisons were performed for rectal infection results between baseline (V2) and V6 (day 56), V10 (day 112), V13 (day 140), V14 (day 168), V15 (day 196), and V16 (day 224). Suppressed rectal explant HIV-1 p24 production was found at all postbaseline (V2) visits compared to V2 for both HIV-1BaL (Clade B) and the G147-1 (Clade C) viruses, where p24 amounts were lower by approximately 0.5–1 log following HIV-1BaL ex vivo challenge and 2 logs following G-147-1 ex vivo challenge in RT (Fig. 3A). In contrast, suppressed ET viral suppression, compared to baseline (V2), was only seen at one time point (V6) and at no time points for VT.
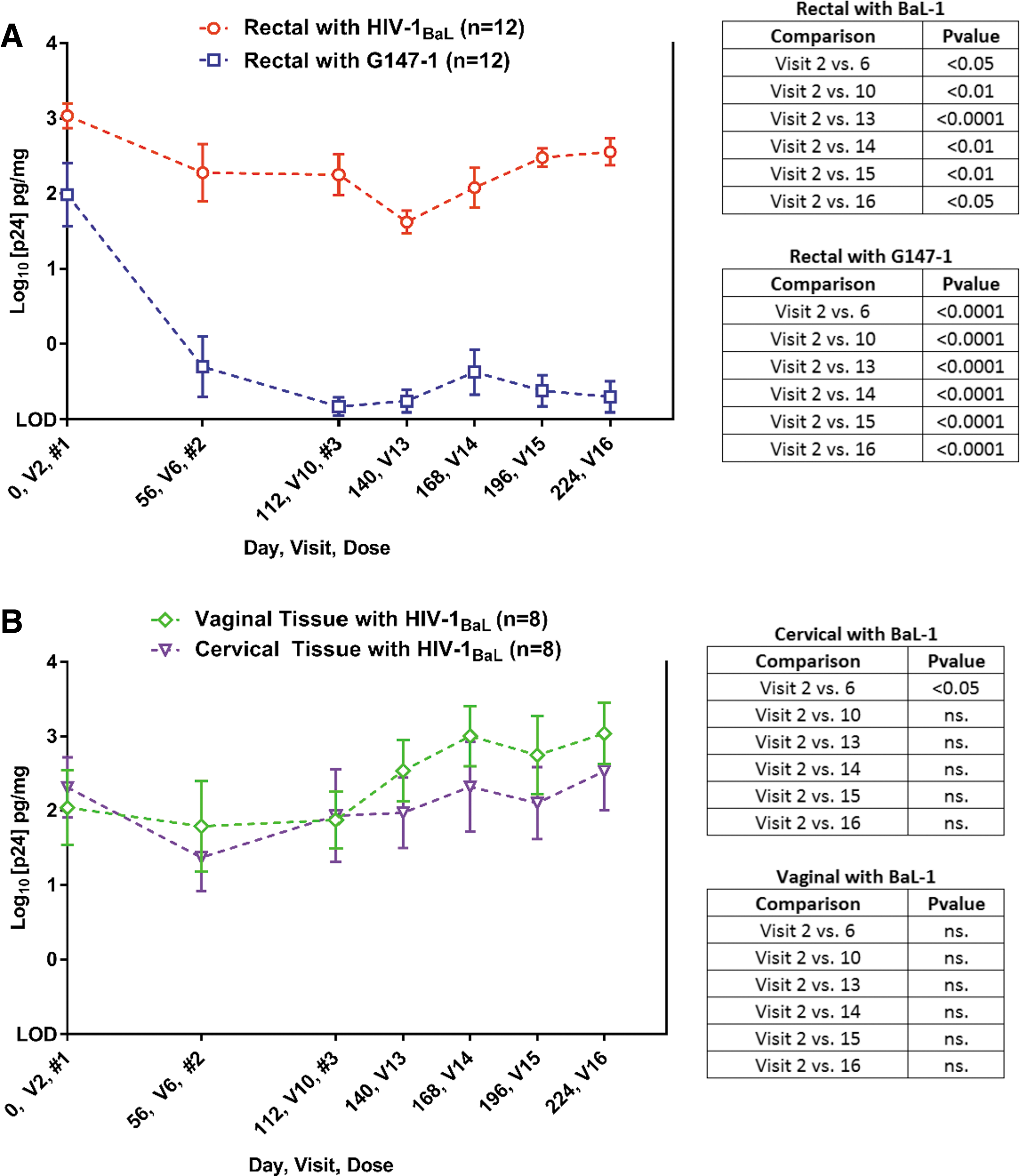
Ex vivo/in vitro explant infection. Tissue explants
PK/PD relationship
Ex vivo infection results in RT, ET, and VT were correlated with paired PK drug levels in the respective tissue across all visits. A linear mixed-effect model was performed to correlate the PK concentrations (i.e., concentration in tissue) with the PD response (i.e., cumulative p24). A significant negative slope supports a finding of drug-mediated virus suppression and was found for RT PK:PD models following ex vivo infection with both BaL-1 and G147-1 for both RT (p < .001; Fig. 4A, B) and plasma (p < .001; Fig. 4C, D) PK models. There were no significant PK:PD relationships found for either ET (Fig. 4E) or VT (Fig. 4F) linear models. The following EC50 parameters (EC50 in ng/mL ±95% CI) were calculated: RT RPV for ex vivo RT infected with either HIV-1BaL: 28.2 (20.0–39.8; Fig. 5A) or HIV-1G147-1: 8.9 (4.3–18.6; Fig. 5B) and plasma RPV for ex vivo RT infected with either HIV-1BaL: 24.5 (17.0–34.7 Fig. 5C) or HIV-1G147-1: 10.5 (5.8–18.6; Fig. 5D).
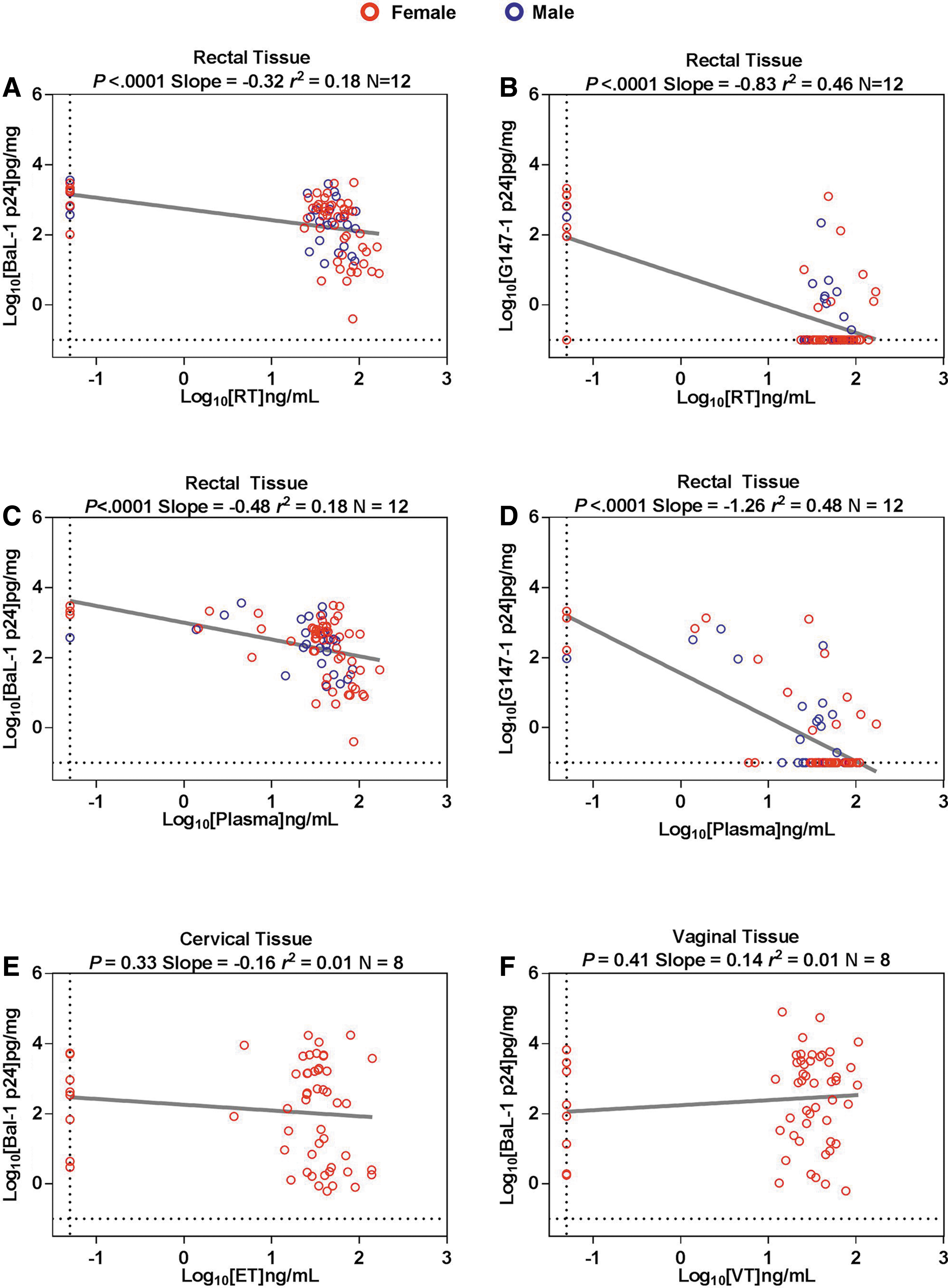
Pharmacokinetic/pharmacodynamic profile of RPV in rectal and cervicovaginal tissue.

EC50 parameters for RPV.
Discussion
Given the challenges associated with long-term adherence to oral antiretroviral therapy, there is growing interest in the development of LA antiretroviral formulations for both the treatment and prevention of HIV infection.
6,27
The MWRI-01 study was undertaken to characterize safety, acceptability, PK, and PD following multiple-dose administration of RPV LA. Single-dose administration of RPV LA has previously been shown to be safe and well tolerated.
9
The most common AE was injection site discomfort, which was mild or moderate, transient, and did not appear to diminish participants' willingness to consider this form of HIV prevention in the future. Although isolated instances of Grade 1 prolongation of the QTc interval was seen in two participants; this was an artifact of the DAIDS toxicity scoring system, which has subsequently been revised.
The behavioral data suggest that study participants were not dissuaded by fear of injection-associated pain and were likely to use the product. Product cost, however, was a significant factor in the perceived likelihood of ‘real world’ uptake. In the HPTN 076 study (NCT02165202) where female participants received six 1,200mg injections of RPV at 8-week intervals, 61% of the women said they would definitely use this form of HIV prevention in the future. 28
Following administration of three IM injections of RPV LA, maximum plasma concentrations of RPV were achieved by 7 days postinjection; however, due to the study's sparse sampling design, it is possible that the true Cmax may have been missed if peak levels occurred between days 7 and 28. Drug accumulation was evident in all anatomical compartments after the second injection of the RPV LA formulation.
Although plasma levels were consistently lower in the male participants compared to the female participants, overall RPV plasma concentrations exceeded the drug's protein-adjusted EC90 throughout the study duration. Moreover, data from the multidose arms support our initial findings from the single-dose study, demonstrating that RPV persists in plasma with drug levels remaining above the EC90 for ≥4 months. 9 Similarly, recent data from the HPTN 076 trial showed RPV plasma concentrations in excess of the EC90 for up to 8 months after a sixth RPV LA injection in female subjects receiving an equivalent regimen (HPTN 076 included an initial 4-week dosing period of daily oral RPV). 28 In this study, tissue levels were variable, but in general, RPV concentrations in RT were higher than in the cervical or VT of female participants: for example, for concentrations at 112 days, median tissue-to-plasma ratios were 1.33 for RT and ∼0.75 for ET/VT.
As with our previous SD data, ex vivo infection rates were lower in RT, showing evidence of drug-mediated virus suppression, and this was sustained out to 224 days after RPV LA administration. Baseline levels of Clade C HIV infection were slightly lower than for the Clade B HIV-1BaL virus (Fig. 3). However, the degree of RT infection suppression was significantly greater (p < .05) for Clade C compared to Clade B infection. There were no significant reductions in virus infection in either cervical or VTs, even though drug levels were detectable in both compartments. This was despite a modest, but significant, increase in tissue levels of RPV seen with repeat dosing. Divergent results from the rectal and cervicovaginal tissue models may reflect the lower concentrations of RPV seen in the ET and VT and/or differential PK requirements to suppress explant infection in either compartment. 29 Furthermore, lack of protection in the ET and VT could also attribute to loss of RPV from the explant tissue, which has been demonstrated by Dezzutti et al. (8% of the total amount of RPV was quantified from the total amount added to basolateral cultures of ET). 29
One limitation of injectable LA PrEP is that once the products have been injected, they cannot be removed and so any idiosyncratic AEs associated with their use will be difficult to manage. In addition, the long terminal PK tail associated with LA PrEP has raised concerns about the possibility of antiviral resistance emerging in individuals who are infected during this period. 30 Current trials evaluating cabotegravir LA are using an oral run-in period to try and identify individuals who might develop product-related AEs and up to 18 months of oral PrEP with emtricitabine/TFV disoproxil fumarate once the LA PrEP is discontinued. It is unclear whether this strategy will be successful and so there is now increasing interest in developing implantable devices that could be easily removed. Ideally, these products would gradually release the antiretroviral drug over a period of months, if not years. This profile limits the number of suitable antiretroviral candidates.
In this Phase 1 study, we have demonstrated that 3 IM doses of RPV LA are safe and acceptable. Moreover, exposure to three doses of RPV LA was associated with plasma RPV levels that were sustained above the protein-adjusted IC90 between doses and with viral suppression in colorectal, but not cervicovaginal tissue.
Footnotes
Acknowledgments
The authors would like to thank the participants for their commitment to the MWRI-01 study. The MWRI-01 study was funded by a grant from the Bill and Melinda Gates Foundation (OPP1045325).
Authors' Contributions
I.M. designed the study protocol with assistance from R.D.C. and K.Z.A. Laboratory assays were run by A.S., J.E., and C.S. and overseen by R.M.B and C.S.D.; D.B., L.E., D.E., and S.K were responsible for the analysis of all PK samples; R.S. and J.E.E designed, conducted, and analyzed the behavioral component of the study; K.Z.A. and N.R.-H. analyzed and interpreted the data; P.W. provided access and regulatory support for the provision of RPV LA; and R.D.C. and I.M. wrote the article. All authors have read and commented on the final article.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.