
Abstract
Human immunodeficiency virus type 1 (HIV-1) is a major causative agent of acquired immune deficiency syndrome. Subtype C (HIV-1C) is the most prevalent HIV-1 subtype worldwide. Although it is highly prevalent in Nepal, genotypic information on Nepalese HIV-1C is limited. We herein investigated the origin and dynamics of HIV-1C in Nepal. Nearly full-length sequencing of Nepalese HIV-1C strains and phylogenetic analyses were performed. The results obtained showed that Nepalese HIV-1C is closely related to the Indian and southern African strains and the introduction of HIV-1C into Nepal was estimated to be in the mid-1980s. These results suggest that multiple HIV-1C strains entered Nepal in the mid-1980s, and this was followed by a marked increase in the number of infection cases for the next decade. These results reflect the current transmission dynamics of HIV-1C strains in Nepal and provide valuable information for HIV monitoring and vaccine development.
Human immunodeficiency virus (HIV) is a lentivirus belonging to the family of Retroviridae and is a causative agent of acquired immune deficiency syndrome (AIDS). In 2017, 1.8 million people were newly infected with HIV and the total number of people living with HIV reached 36.9 million worldwide. 1
In recent years, antiretroviral therapy (ART) has been the standard therapy for HIV infection. ART enables HIV-infected individuals to have low viral loads and prolongs life expectancy. 2 Despite the success of ART for both the treatment and prevention of HIV infection, a safe and effective vaccine will be needed to achieve a practical and durable end to the global HIV pandemic. 3 However, the development of an effective HIV vaccine has not yet been achieved. 4 One of the key scientific hurdles is the extensive genetic diversity of the virus. HIV diversity plays a central role in the HIV pandemic; therefore, continuous global molecular epidemiological surveillance is of importance.
Phylogenetic analyses have been the key for identifying the patterns and mechanisms of natural selection, which is essential for effective HIV vaccine development. They also provide information on HIV transmission dynamics essential for effective HIV control. Phylogenetic analyses are common practices in many HIV/AIDS research programs, 5 and provide many insights and novel questions related to HIV biology.
HIV has been divided into two types: HIV type 1 (HIV-1) and type 2 (HIV-2). HIV-1 infection is responsible for the majority of AIDS cases worldwide, and has been classified into four groups: M (major), O (outlying), N (new or non-M, non-O), and P (pending). Viruses in group M have been further classified into many subtypes and circulating recombinant forms (CRFs). 6 Among the various subtypes and CRFs, HIV-1 subtype C (HIV-1C) is predominant and associated with >50% of all HIV-1 infection cases. 7 HIV-1C is predominant in India and neighboring countries, as well as in southern African countries. 6,7 Furthermore, the prevalence of HIV-1C is increasing in China and eastern Africa. 8 Owing to the high prevalence of HIV-1 subtype B in developed countries, research on the natural history of HIV-1 and health technology improvements have largely focused on this variant. However, effective vaccine design requires further studies on HIV-1C.
In Nepal, which is located between China and India, HIV-1C accounts for nearly 50% of all infection cases. 7,9 In 2017, the total number of people living with HIV reached 31,000. 10 However, limited information is currently available on the genetic diversity of HIV-1C in Nepal. In this study, we performed sequencing and reconstruction of the nearly full-length viral genome to elucidate the phylogenetic relationship between HIV-1C in Nepal and related strains in other countries. We also conducted molecular dating on Nepalese HIV-1C to estimate the time at which it was introduced into Nepal.
Forty blood samples were collected at 10 health centers in Kathmandu, Makwanpur, and Chitwan, Nepal, between 2015 and 2016. Detailed information on study participants was provided in our previous study.
11
Before blood collection, ethical clearance was obtained from the Institutional Ethics Committees of Kobe University Graduate School of Health Sciences (Ethical approval no. 434) and the Nepal Health Research Council (Ethical approval reg. no. 274/2015). DNA was extracted from whole blood samples using a Genomic DNA Mini Kit (Thermo Fisher Scientific, Carlsbad, CA). HIV-1 genomes amplified from 35 blood samples were classified as HIV-1C.
11
The nearly full-length HIV-1C genome was amplified in two fragments. The 5′ terminal half of the HIV-1 genome [4,307-base pair (bp), (first fragment)] and the 3′ terminal half of the genome (4,128-bp, second fragment) were individually amplified by nested PCR using the Expand Long Template Enzyme system (Roche Diagnostics, Indianapolis, IN) and following primer sets. To amplify the first fragment, msf12b; 5′-AAATCTCTAGCAGTGGCGCCCGAACAG-3′ [corresponding to nucleotide (nt) 623–649 of the HIV-1 reference strain, HXB2 (GenBank accession no. K03455)], and DRIN02; 5′-CCTGTATGCAGACCCCAATATG-3′ (nt 5243–5264) were used for first PCR and f2nst; 5′-GCGGAGGCTAGAAGGAGAGAGATGG-3′ (nt 769–793) and DRIN04; 5′-TAGTGGGATGTGTACTTCTGAAC-3′ (nt 5195–5217) were used for nested PCR. In addition, to amplify the second fragment, DRIN01; 5′-CAGACTCACAATATGCATTAGG-3′ (nt 4039–4060) and UNINEF 7′; 5′-GCACTCAAGGCAAGCTTTATTGAGGCTT-3′ (nt 9605–9632) were used for first PCR and DRIN05; 5′-TGGCATGGGTACCAGCACACAA-3′ (nt 4146–4168) and nefyn05; 5′-GTGTGTAGTTCTGCCAATCAGGGAA-3′ (nt 9157–9181) were used for nested PCR.
12,13
PCR conditions were as follows. In the first PCR amplification of the first fragment, 1 cycle at 94°C for 2 min for denaturation; 10 cycles at 94°C for 10 s for denaturation, at 55°C for 30 s for annealing, and 6 min for extension; 25 cycles at 94°C for 10 s for denaturation, at 55°C for 30 s for annealing, and 6 min 20 + 20 s cycle elongation for each successive cycle at 68°C for extension; and a final extension cycle at 68°C for 10 min were performed. In the nested PCR amplification of the first fragment and the first and nested PCR amplification of the second fragment, annealing temperatures were changed to 50°C, 50°C, and 55°C, respectively. Sanger sequencing of successfully amplified first and second fragments was then performed by Macrogen Japan (
The nearly full-length sequences of four HIV-1C strains, NP13, NP20, NP32, and NP36, were obtained. In addition, the gag and pol genes of the HIV-1C strain, NP15 were successfully sequenced. These nucleoside sequences are available under GenBank accession numbers MK493076–MK493080. All available HIV-1C nearly full-length sequences were downloaded from the HIV Sequence Database (
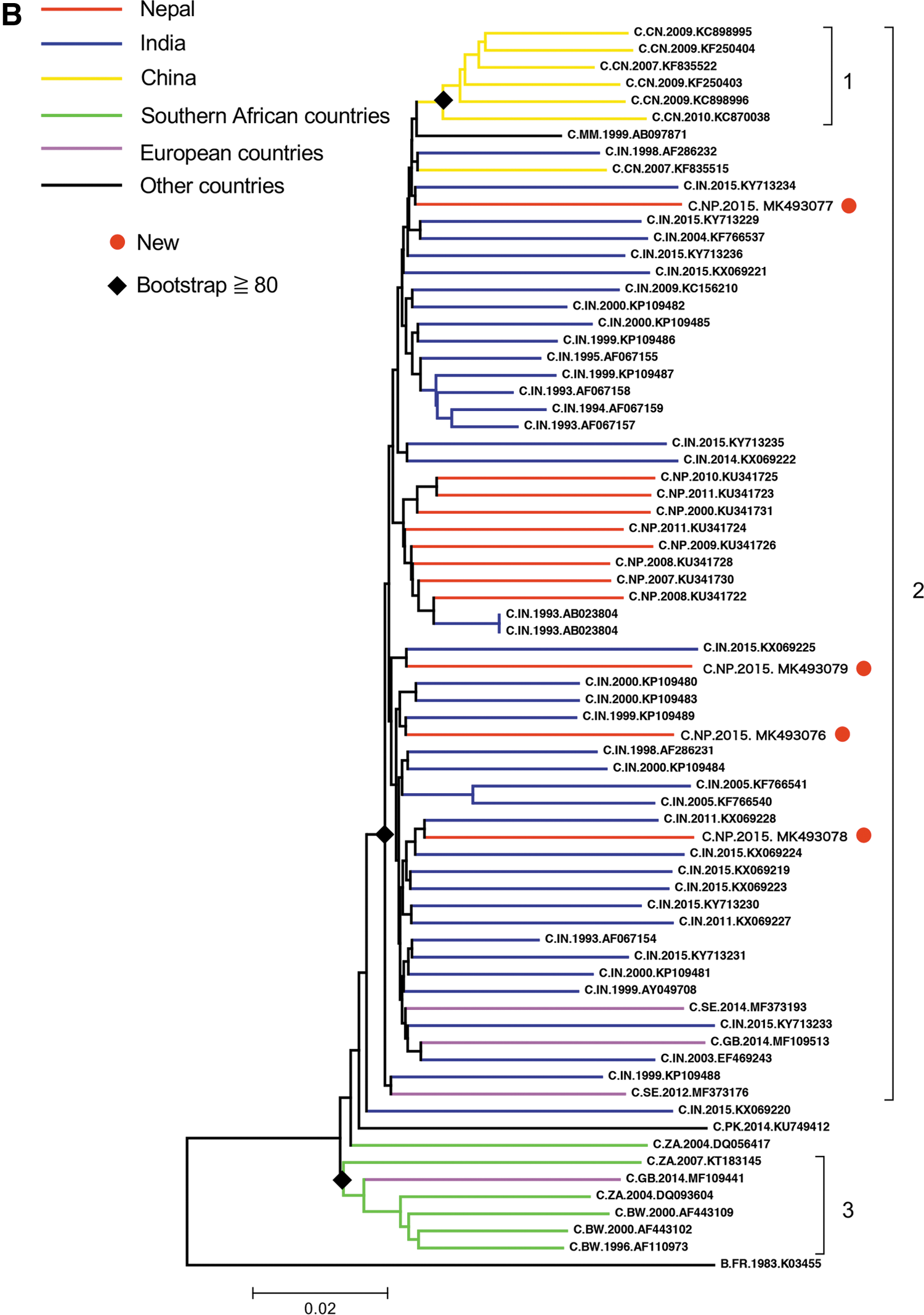
The final data set included a total of 685 sequences, including 12 Nepalese sequences (among which 4 were newly sequenced in this study and 8 were downloaded sequences). Sequences were from 4 continents and 20 countries: Asia (6 countries), Africa (9 countries), the Americas (2 countries), and Europe (3 countries) (Table 1). The NJ tree of 685 HIV-1C sequences is shown in Figure 1A. The phylogenetic tree shows the presence of two clusters: one for the majority of Chinese strains (99% bootstrap support) and one for the majority of Brazilian strains (95% bootstrap support) (Fig. 1A). No Nepalese strains were observed in these clusters. Furthermore, there was no unique cluster of Nepalese strains. The four new Nepalese sequences were closely related to Indian strains (Fig. 1A).

Phylogenetic analyses of four nearly full-length Nepalese HIV-1C sequences.
Number of HIV-1C Strains from Different Countries Included in This Study
HIV-1C, HIV-1 subtype C.
To elucidate and visualize the relationship between Nepalese HIV-1C sequences in more detail, another NJ tree was generated with the top 67 sequences closely related to the 4 new Nepalese sequences. There were two small clusters and a major cluster. Cluster 1 (Chinese strains) and cluster 3 (southern African strains) were each supported by 99% and 94% bootstrap support, respectively (Fig. 1B). The four new Nepalese sequences fell into a major cluster (cluster 2: 80% bootstrap support), including strains from India, China, and European countries (Fig. 1B). The aforementioned analysis suggested that HIV-1C strains in Nepal were introduced from India.
We then performed a molecular clock analysis using the gag gene to infer the epidemic histories of HIV-1C in Nepal. This gene is suitable because it does not vary as much as the env gene and is not under selective pressure by antiretroviral drugs, such as the pol gene. The gag sequences of the top 50 strains that have high similarity to each of the 5 new Nepalese sequences were downloaded from the HIV Sequence Database using a BLAST search. To maximize the sampling window and increase the amount of information needed to estimate the molecular clock rate, we selected some of the earliest sampled sequences available in the database. The temporal range of sampling in the reference data sets was between 1993 and 2015. We performed a preliminary phylogenetic analysis using the NJ method with all downloaded sequences (data not shown). We then selected sequences that were closely related to new Nepalese sequences and assembled a data set with 5 new Nepalese sequences and 64 close relatives. A phylogenetic analysis was performed using a Bayesian approach as implemented in BEAST version 1.8.4 using the general time reversible (GTR) model of nucleotide substitution, assuming Γ-distributed rates among sites and invariable sites. We used the uncorrelated lognormal relaxed clock model with Tip-Dates and Bayesian skyline as coalescent tree priors. Two separate Markov chain Monte Carlo (MCMC) runs were made for 108 generations with a burn in of 107. MCMC was sampled every 103 generations. No additional priors were used in the analysis. We assessed the convergence of the Markov chain [effective sample size (ESS) >200] using the program Tracer v1.6.0. The final tree was compiled in TreeAnnotator and edited in FigTree. The Bayesian tree of the 69 HIV-1C gag sequences is shown in Figure 2. Nepalese sequences were interspersed with Indian and southern African strains, and no unique cluster was observed (Fig. 2). Thus, this analysis showed that HIV-1C was introduced into Nepal on multiple occasions. The 95% highest posterior density intervals of the introduction dates of five Nepalese HIV-1C gag sequences are shown in Figure 3. The median date estimates of their introductions were similar. As a result, the date of HIV-1C introduction into Nepal was estimated to be in the mid-1980s (Fig. 3).
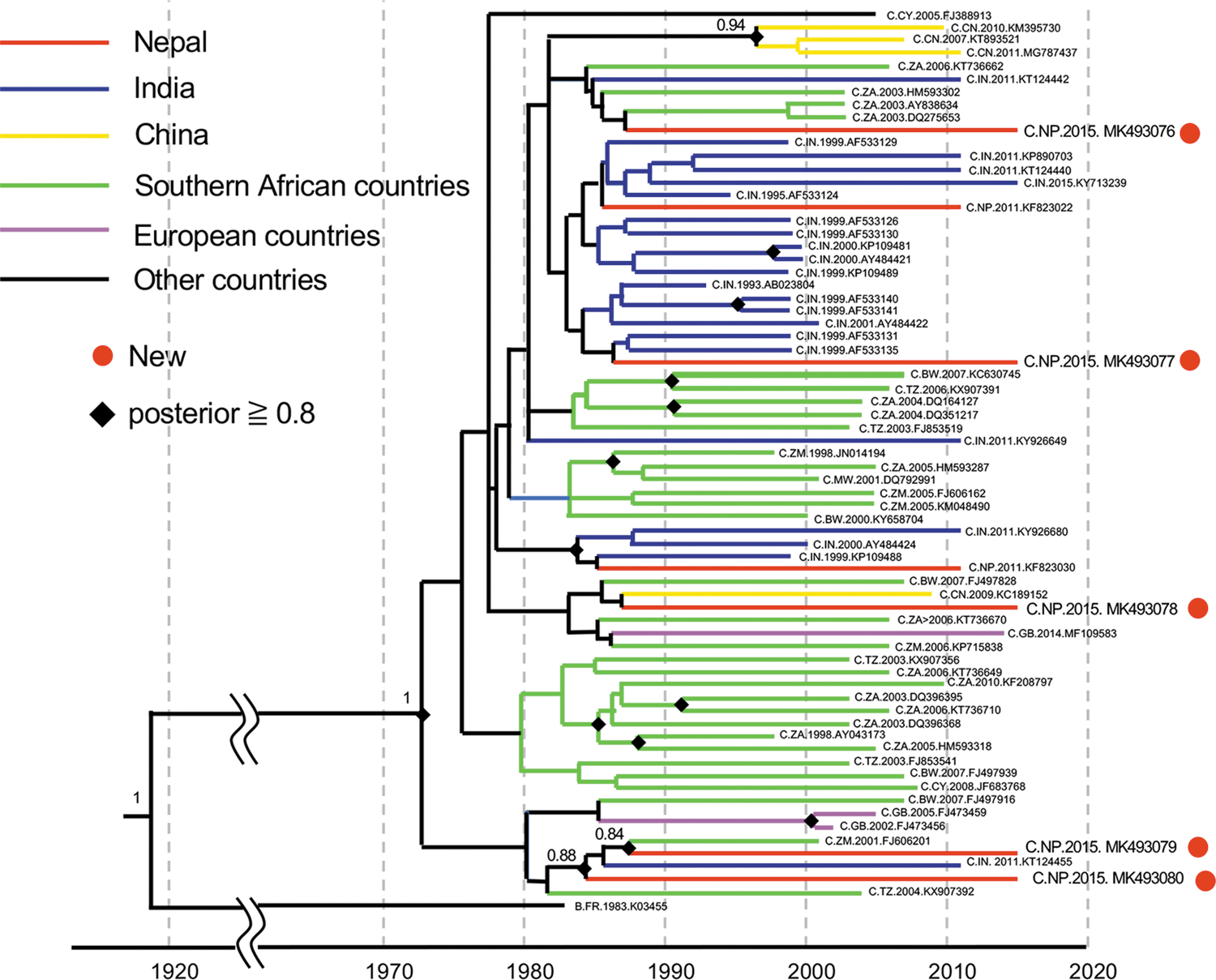
Bayesian tree with the timescale of 69 HIV-1C gag sequences. A maximum clade credibility tree with a timescale was obtained using the gag gene sequences (nt 1186–1879, 694-bp) of 69 HIV-1C strains. The tree was obtained using a Bayesian approach as implemented in BEAST version 1.8.4 using the GTR model of nucleotide substitution, assuming Γ-distributed rates among sites and invariable sites. Color images are available online.
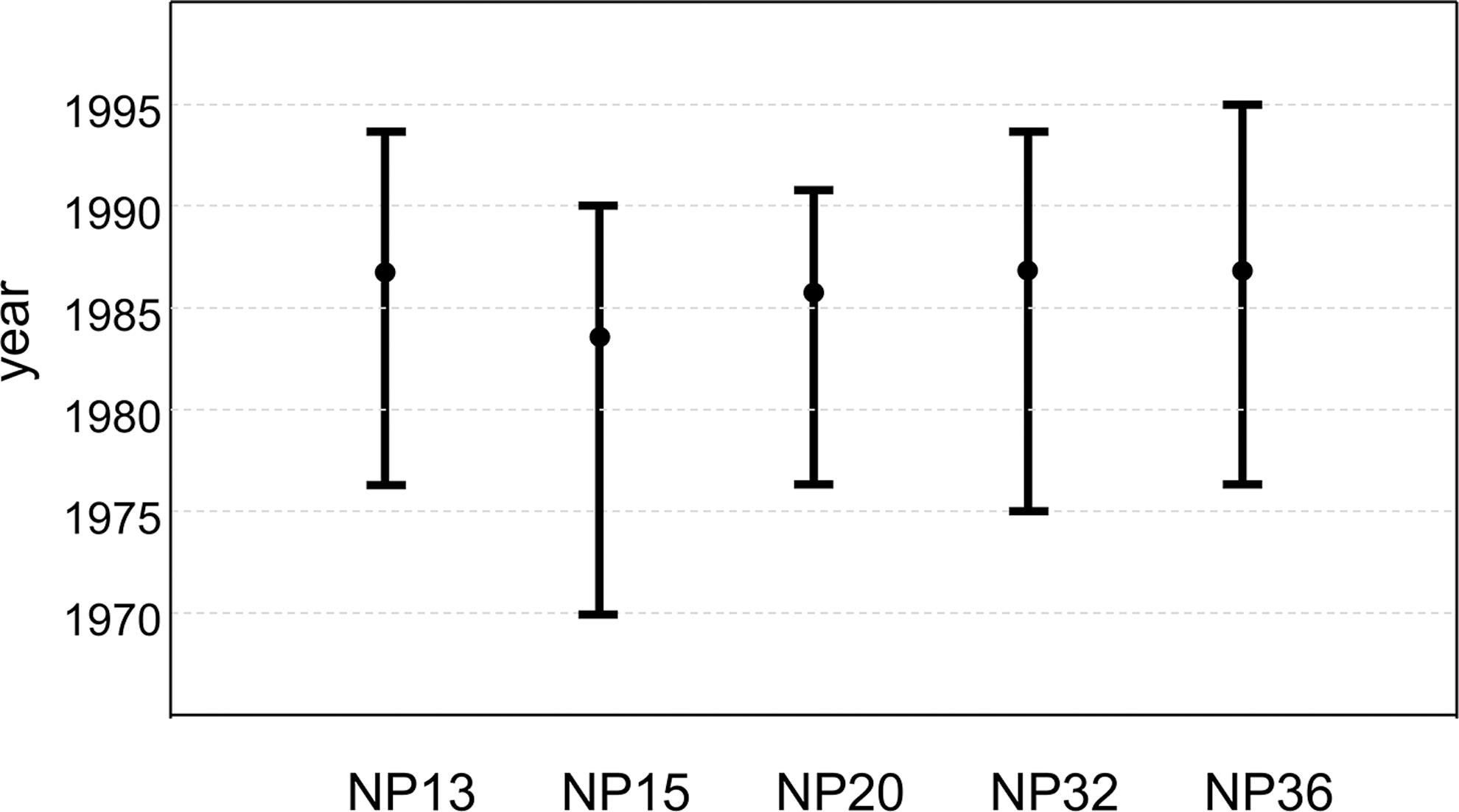
Dating the subtype C epidemic in Nepal. Coalescent-based estimations and 95% highest posterior density intervals of the introduction dates of five new Nepalese sequences were estimated based on the results shown in Figure 2.
In this study, we analyzed the geographical origins and introduction dates of Nepalese HIV-1C to clarify its evolutionary history. The phylogenetic analysis of all available nearly full-length reference sequences on the database provided an adequate representation of global HIV-1C diversity. The results obtained showed greater phylogenetic similarities in Nepalese sequences to Indian strains than to Chinese strains (Fig. 1A). However, Nepalese sequences did not form a unique cluster. We then conducted a detailed NJ analysis on 71 sequences closely related to Nepalese strains. There were three clusters in the phylogenetic tree (Fig. 1B), and Asian strains formed a unique cluster that differed from southern African strains. Nepalese strains were closely related to Indian strains, but did not form a unique cluster. These results suggest that HIV-1C was introduced into Nepal on different occasions.
In contrast to the results of the phylogenetic analysis using nearly full-length sequences, the analysis of 69 HIV-1C gag sequences provided an additional insight into the introduction of HIV-1C into Nepal. The phylogenetic tree showed that Nepalese strains were closely related to southern African strains (Fig. 2). The discrepancy in the phylogenetic relationship between nearly full-length and gag sequences suggests the recombination of two or more HIV-1 genetic fragments in the genome. Bhusal et al. showed that the key risk groups for HIV-1 infection were male labor migrants and migrant sex workers in Nepal. 9 In addition, Neogi et al. reported that HIV-1C was introduced into India by migration from southern Africa. 14 These two studies suggest that the key risk groups for HIV-1 infection in Nepal were infected with both strains from India and southern Africa. Thus, the recombination of HIV-1C from India and southern Africa may have occurred. Further continuous surveillance is required to monitor the emergence of new CRFs. Our estimates in Figure 3 suggest that HIV-1C was introduced into Nepal in the mid-1980s, ∼10–15 years before a marked increase in the number of infections. 15 The strong growth of HIV-1C in southern Africa in the 1980s and the introduction of HIV-1C into India by migration from southern Africa confirmed our estimates. 14
In summary, our evolutionary reconstructions suggest that multiple HIV-1C entered Nepal from India and southern Africa in the mid-1980s, and this was followed by a marked increase in the number of infections for the next decade. 15 These results reflect the current transmission dynamics of HIV-1C strains in Nepal and provide valuable information for HIV monitoring and vaccine development.
Footnotes
Acknowledgments
This study was supported, in part, by the field research program, L3.TAP15, by the Research Center for Urban Safety and Security (RCUSS), Kobe University; a research grant by the Kobe University Graduate School of Health Sciences; the program of the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from the Ministry of Education, Culture, Sport, Science and Technology in Japan, and the Japan Agency for Medical Research and Development (AMED). The article was proofread by Medical English Service, Kyoto, Japan.
Sequence Data
Nucleoside sequences are available under GenBank accession numbers MK493076–MK493080.
Author Disclosure Statement
No competing financial interests exist.