
Abstract
HIV-1 intersubtype B/C recombinants were more commonplace reported in high-risk populations in Yunnan province, China. In this study, three unique (B/C) recombinant isolates (2015YL02, 2018YL07, and SN18015) were identified from patients infected with HIV-1 through sexual transmission in Shaanxi province. Phylogenetic and bootscan analyses showed that three recombinants comprised HIV-1 subtype B and subtype C. The recombinant structures revealed that the near full-length sequence of 2015YL02 shared an identical mosaic structure with XC2014EU20, which was isolated from Sichuan province. And two similar breakpoints were observed between 2018YL07 and CRF08_BC in pol gene. The arising B/C recombinant forms enrich evidence of the HIV-1 genetic diversity among sexually transmitted populations and suggest that continuously monitoring HIV-1 molecular epidemiology is needed in Shaanxi province.
China has experienced a great change of HIV/AIDS pandemic, from blood transmission and injecting drug users (IDUs) to sexual transmission. As sexual contact is the dominant transmission mode and infection cases continue to increase, 1 high complexity of genotypes distribution was observed in HIV-1 pandemic of China. 2 A wide variety of HIV-1 genotypes, circulating recombinant forms (CRFs), and unique recombinant forms (URFs) were prevalent in high-risk populations. CRF01_AE, CRF07_BC, subtype B/B’, CRF08_BC, and subtype C were predominant strains in China. 3 Studies emphasized that coexisting of subtypes B and C gives rise to the generation of various CRFs_BC and URFs_BC. 4 –6 CRF07_BC and CRF08_BC identified in Yunnan province, 7 were two predominant CRF_BC strains in China. In this study, we report three unique recombinant strains composed of genomes B and C from sexually transmitted HIV infections in Shaanxi province.
Shaanxi province, including 10 prefectures, is an administrative province in the northwestern part of China and neighbors Henan province at the eastern border. A total of 98.2% of newly diagnosed HIV/AIDS cases of 2018 were infected through sexual exposure. 8 The proportion of the patients, older than 60 years, has increased four times compared with that 5 years ago. And the number of newly reported cases was growing rapidly in the northern part of Shaanxi province. The three HIV-positive patients in this study are SN18015 living in Baoji prefecture and the other two, 2015YL02 and 2018YL07, living in Yulin prefecture. Baoji is located in the western part of Shaanxi, with a population of 3,781,000 and an area of 18,100 square kilometers. Yulin, located in the most northern part of Shaanxi, has a population of 3,700,000 and an area of 43,578 square kilometers (Fig. 1).

Map depicting study sites.
The whole blood sample of SN18015 was collected from the surveillance of transmitted drug resistance among nontreatment people who were newly diagnosed with HIV-1 infection in 2018. Plasma samples of 2015YL02 and 2018YL07 were collected from the molecular epidemiologic survey of HIV/AIDS cases resident in Yulin prefecture. The demographic information of the patients is summarized in Table 1. All patients gave signed written informed consent before sample collection. This study was reviewed by the Institutional Research Ethics Community of the Shaanxi Center for Disease Control and Prevention.
Demographic Information of Three Individuals Infected with HIV-1 Unique (B/C) Recombinant Strains
The patient, 2015YL02, was dead in February 2015 because of the opportunistic infections.
The near full-length genome (NFLG) was amplified and sequenced as previously described.
9
The NFLG sequences were codon aligned by the Gene Cutter tool with subtype reference sequences (all HIV-1 group M, CRF01_AE, CRF07_BC, CRF08_BC, and other CRFs_BC) as well as URFs_BC identified in China downloaded from the Los Alamos National Database, and then edited manually by Bioedit 7.0. Phylogenetic tree analyses were conducted using the neighbor-joining method based on the Kimura two-parameter model with 1,000 bootstrap replicates by Mega 5.0. Recombinant patterns were first determined by the Recombinant Identification Program (RIP) and the jumping profile hidden Markov model (jpHMM) (
In the phylogenetic tree (Fig. 2A), three NFLGs of this study formed distinct monophyletic clusters, distantly from all known HIV-1 subtypes/CRFs. The NFLG of 2015YL02 clustered with XC2014EU20, 10 a URF_BC strain isolated from a male who was infected through heterosexual sex in Sichuan, with a bootstrap value 100%. The recombinant pattern analyses displayed three NFLGs and were all composed of genomes B and C (Fig. 2B), with subtype B (RL42), subtype C (95IN21068), and CRF01_AE (CM240) as references. Two BC recombinants obtained from Dehong, Yunnan, 07YNLC22 and 09YNYJ217010, were close to the NFLG of SN18015 in the phylogenetic tree. They were chosen to implement the comparison of recombinant structures with SN18015. The result of bootscanning plots showed that the three NFLGs shared a similar breakpoint in the env region (Fig. 2B).
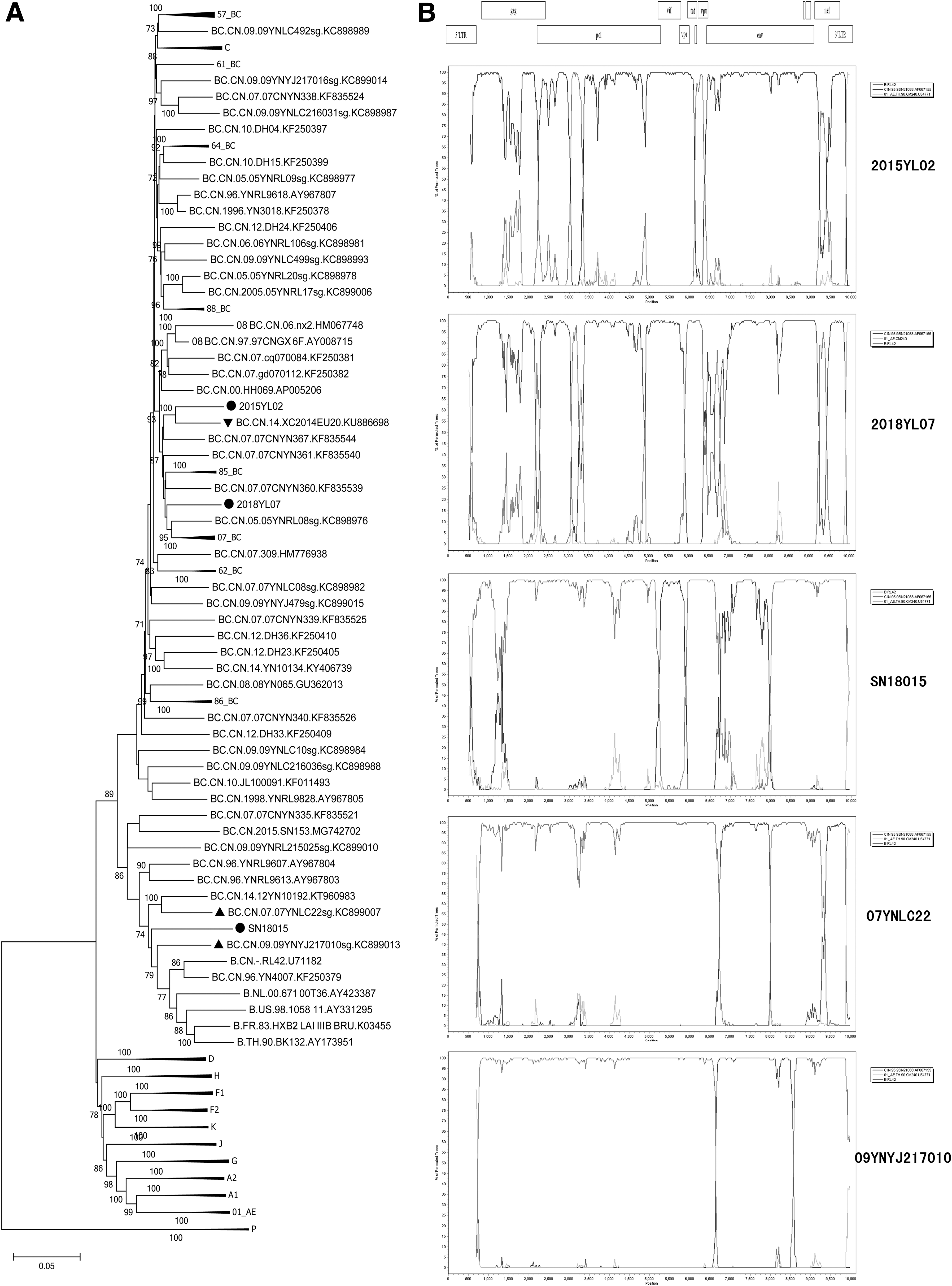
The phylogenetic tree and the bootscanning plots of the novel identified URFs_BC.
The mosaic recombinant structures of the three NFLGs are as follows: IC (633-2872 nt), IIB (2873-3205 nt), IIIC (3206-6053 nt), IVB (6054-6234 nt), VC (6235-8849 nt), VIB (8850-9038 nt), and VIIC (9039-9602 nt), 2015YL02; IC (642-2021 nt), IIC (2202-2866 nt), IIIB (2867-3163 nt), IVC (3164-5786 nt), VB (5787-6192 nt), VIC (6586-8858 nt), VIIB (8859-9047 nt), and VIIIC (9048-9602 nt), 2018YL07;. IB (633-5105 nt), IIC (5106-5786 nt), IIIB (5787-6545 nt), IVC (6546-7687 nt), and VB (7688-9597 nt), SN18015 (Fig. 3). Subregion phylogenetic analyses were used to confirm the genetic origins of each segment. The subtype B regions might derive from subtype B-Thai, reference RL42 from China. The parental origin of genome C regions clustered with India C lineage (Supplementary Fig. S1). Some of the segments were probably too short to determine their origin. As is shown in Figure 3, six breakpoints are similar between the genomic maps of 2015YL02 and XC2014EU20. And the recombinant structures of both strains composed of three segments of genome B inserted into the pol, vpu, and nef regions of the subtype C backbone, which is quite similar to CRF85_BC. 11 It is necessary to pay attention to this URF, since it may be a potential CRF. And two similar breakpoints are also observed between 2018YL07 and CRF08_BC in the pol gene (Fig. 3).

The recombinant maps of near full-length sequences of 2015YL02, XC2014EU20, 2018YL07, CRF08_BC, and SN18015, respectively. The mosaic structures created using the Recombinant HIV-1 Drawing Tool (
Studies characterized the considerable recombinant prevalence of URFs_BC circulating among IDUs 4 and heterosexual populations 6 in Dehong prefecture, Yunnan. Our study reports that the URFs_BC strains also arise among sexually transmitted populations in the northwestern part of China. Shaanxi is one of the sexually driven epidemic provinces in China. Second-generation recombinant forms were already reported among local sexually transmitted groups. 9,11 Hence, the need for surveillance B/C recombinant genotypes should be drawn attention in the work of molecular epidemiologic investigation.
In addition, studies noticed that the identical recombinant breakpoints were being shared among different recombinant patterns of URFs_BC. 4 –7 In this study, similar recombinant breakpoints, supposed to be hotspots, were observed between URFs and CRFs. This provides evidence to the direct evolutionary relationship of BC recombinants. 7 It is interesting to note that BC recombinant patterns, composed of genome B variable regions inserted into subtype C parental backbone, were more likely to be established as CRFs, such as CRF07_BC, CRF08_BC, CRF57_BC, CRF61_BC, CRF62_BC, CRF64_BC, and CRF85_BC. However, we should be aware that the prevalence of BC recombinant forms may be underestimated. Currently, the amplification of NFLG of URF is only conducted if the inconsistent genotypes or recombinant breakpoints are found among pol, env, and gag gene segments. It is suggested that, along with the surveillance of genotyping HIV-1, harvesting more genetic information besides gene fragments will help us in the study of molecular epidemiology patterns.
Sequence Data
The NFLG sequences of 2015YL02, 2018YL07, and SN18015 are available in GenBank under the accession numbers MK792287–MK792289.
Footnotes
Author Disclosure Statement
The authors declare no competing financial interests.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.