
Abstract
The human T cell lymphotropic virus type 1 (HTLV-1) infects 5 to 10 million individuals and remains without specific treatment. This retrovirus genome is composed of the genes gag, pol, env, and a region known as pX. This region contains four open reading frames (ORFs) that encode specific proteins. The ORF-I produces the protein p12 and its cleavage product, p8. In this study, we analyzed the genetic diversity of 32 ORF-I sequences from patients with different clinical profiles. Seven amino acid changes with frequency over 5% were identified: G29S, P34L, L55F, F61L, S63P, F78L, and S91P. The identification of regions where the posttranslational sites were identified showed a high identity among the sequences and the amino acid changes exclusive of specific clinical profile were found in less than 5% of the samples. We compare the findings with 2.406 sequences available in GenBank. The low overall genetic diversity found suggested that this region could be used in the HTLV-1 vaccine development.
The human T cell lymphotropic virus type 1 (HTLV-1) was the first described human retrovirus. 1 It is estimated that 5–10 million people are infected with HTLV-1 in the world, and although this infection is endemic in different geographic regions, it still remains without effective therapeutic methods. 2 It is known that most patients infected with HTLV-1 do not develop clinical manifestations, but this retrovirus is the etiologic agent of infective dermatitis associated to HTLV-1 (IDH), HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), and adult T cell leukemia/lymphoma (ATLL), among others. 3 –5 The major barriers for the development of HTLV-1 therapeutic vaccine is the comprehension why some individuals develop pathological processes while others remain asymptomatic and what is the best way to prevent viral persistence and infectivity.
A recent research demonstrated that the persistence of HTLV-1 infection is influenced by the expression of p12 and p8 proteins, encoded from the open reading frame (ORF)-I of the pX gene region. This study suggests that some natural ORF-I mutations alter the expression of the p12 and p8 proteins and that equivalent concentrations of both are necessary to prevent recognition and lysis of HTLV-1 infected cells by cytotoxic T cells. 6 We previously suggested that some of these natural ORF-I mutations might influence the proviral load and clinical manifestation of HAM/TSP. 7
Considering the influence of the HTLV-1 ORF-I expression on the course of infection, this study aims to evaluate whether this region could be used as a target for the development of a therapeutic vaccine through the analysis of ORF-I genetic diversity.
In the first stage, we analyzed samples from 32 patients with defined clinical profile: 6 from patients with HAM/TSP, 6 from ATLL patients, 14 from asymptomatic patients, and 6 samples from patients with IDH. The clinical classification was carried out by medical experts according to World Health Organization (WHO). All samples were anonymized and informed consent was written and obtained from each subject. These samples were obtained from Center of HTLV-1 Bahia School of Medicine and Public Health—Salvador, Bahia, Hospital Complex Prof. Edgar Santos—Salvador, Bahia and from Hemocentro—Ribeirão Preto, São Paulo, and the research was approved by the Ethics Committee of the Centro de Pesquisa Gonçalo Moniz/Fiocruz (N0 377/2012).
In the second step, a search of the HTLV-1 ORF-1 sequences available on GenBank was performed to compare the results obtained from the sequences of our patients with other available sequences.
The peripheral blood mononuclear cells were obtained and DNA was extracted using spin column DNA extraction system (QIAamp DNA Mini Kit; Qiagen). The samples were used for amplification of ORF-I through polymerase chain reaction (PCR), as follows: denaturation (94°C, 3 min) annealing (94°C, 15 s), 65°C (45 s), 72°C (1 min), 35 times of cycle, and a final extension of 72°C for 8 min, with the primers 24+ (5′CGTATCGCCTCCCTCGCGCCATCAGAGTATGCTGCCCAGAACAG3′) and 27− (5′CTATGCGCCTTGCCAGCCCGCTCAGGGTTCCATGTATCCATTTCGGA3′). The amplicons were purified using PureLink PCR Purification Kit (Thermo Fisher Scientific) and sequenced in an ABI Prism 3100 DNA Sequencer (Applied Biosystems Inc., Foster City, CA) using Taq FS Dye (Applied Biosystems) terminator cycle sequencing with the same PCR primers.
The files from the 32 sequences were trimmed, manually edited, and aligned to the HTLV-1 reference sequence ATK-1 (J02029) to generate the consensus sequence of each patient. The final dataset was first submitted to a search for the major natural amino acid changes, identified in at least of 5% of the sequences. Then the minor mutations, found in less than 5% of the sequences, were identified. All these analyses were done with Geneious R6 software. 8 The statistical analyses were performed using Fisher's exact test and a p-value lower than 0.05 was considered statistically significant. Then, we compared our results with ORF-I sequences available in the GenBank. This GenBank dataset was composed of 2.406 sequences, with 1.399 sequences from patients with HAM/TSP, 57 from ATLL patients, 945 from asymptomatic patients and 5 sequences from patients with IDH.
To perform the molecular analysis of the mutations identified, physicochemical analysis was carried out using Network Protein Sequence Analysis (NPS@) (
Seven natural amino acid changes with frequency over 5% were identified within the dataset: G29S, P34L, L55F, F61L, S63P, F78L, and S91P. Among them, five were located in specific motifs and were previously described as mutations that influence the expression profile of the HTLV-1 ORF-I protein product: G29S, P34L, F61L, S63P, and S91P. 6 The L55F (found only in sequences of patients with IDH) and F78L mutations were not described yet. Among the seven mutations identified, only P34L was found with a statistically significant difference in the frequency within the IDH and HAM/TSP groups (p = 0.047) (Table 1). Analysis performed with the GenBank available sequences reinforces these data, exception of the L55F and F78L mutations, found at low frequency.
Frequency of Major Open Reading Frame-I Natural Mutations and Their Respective Motif
Mutation able to change the chemical physical profile.
p = 0.047 between IDH and HAM/TSP profiles.
ATLL, adult T cell leukemia/lymphoma; HAM/TSP, HTLV-1-associated myelopathy/tropical spastic paraparesis; IDH, infective dermatitis associated to HTLV-1; SH3, Src homology 3.
The wild-type and mutated sequences were submitted to physicochemical analysis and only the P34L mutation was able to alter protein profile. The NPS@ analysis suggested that the ORF-I product with a leucine in 34 position was less hydrophilic, flexible, and antigenic than the wild type. The accessibility was also decreased, while the hydropathy and membrane-buried helix profile were slightly increased (Fig. 1).
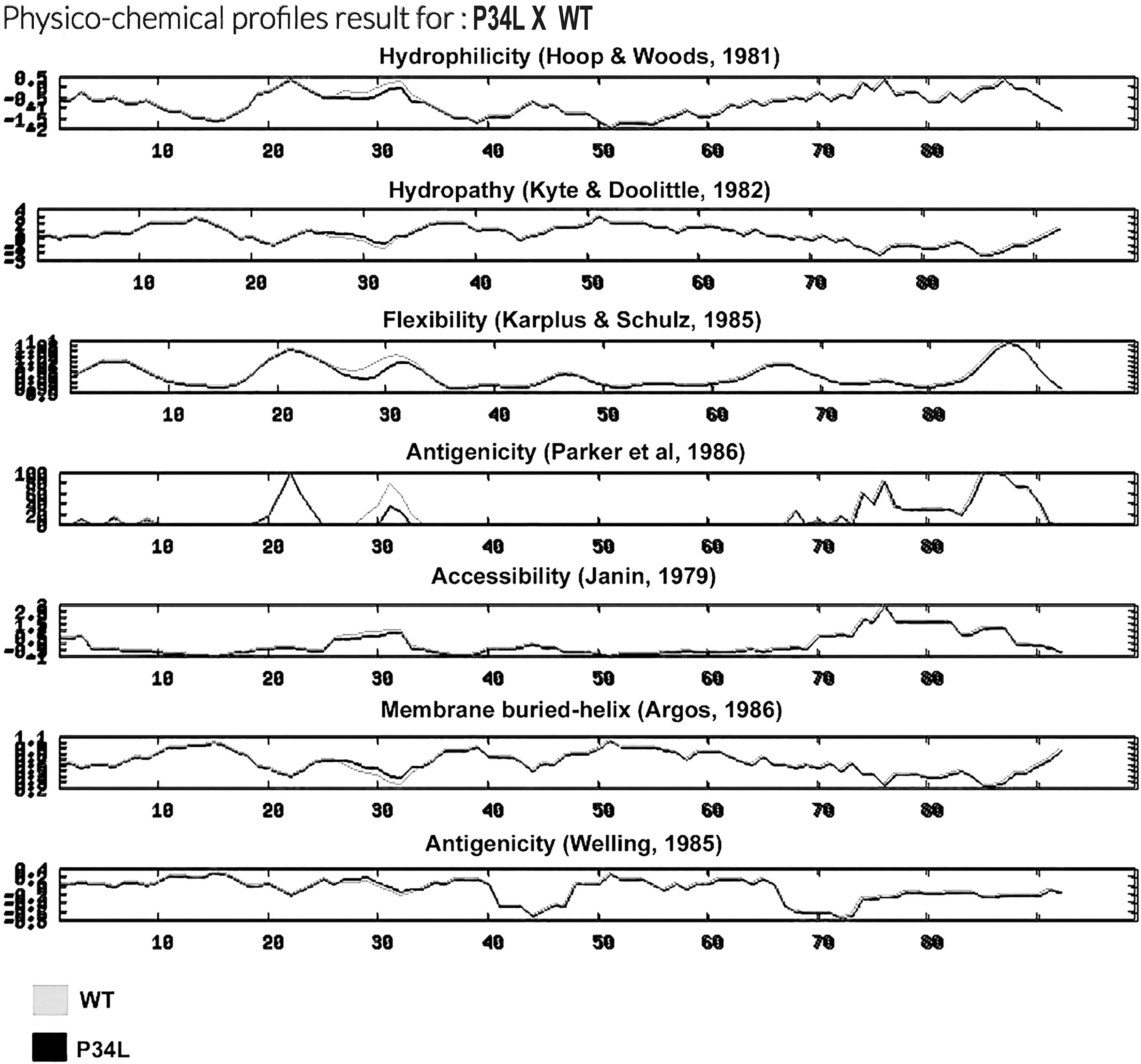
Physicochemical analysis of P34L mutation versus wild type. The graphs are organized as follows: Hydrophilicity; Hydropathy; Flexibility; Antigenicity; Accessibility; Membrane-buried helix; Antigenicity.
To identify if these amino acid mutations were able to create or abrogate potential protein domains, we submitted the 32 ORF-I sequences to the scan PROSITE tool and no changes were observed. All sequences have a casein kinase II phosphorylation site at the 23–26 position and a protein kinase C phosphorylation site at the 75–77 position, which were not altered by the mutations (data not shown).
The analysis of ORF-I sequences revealed 10 mutations found in less than 5% of the samples. Despite being in low frequency, all these mutations have an important characteristic: they are observed only in specific clinical profiles. Six amino acid changes were detected in samples from asymptomatic patients (S7G, P45L, S69G, P73S, R82*, and A96V), two mutations were exclusive of HAM/TSP sequences (C39Y and P86S), while L5I and F84L mutation were identified only in ATLL samples.
In the GenBank dataset, S7G and A96V mutations and the P86S mutation were also found only in asymptomatic individuals and in HAM/TSP patients, respectively.
The overall diversity between sequences from patients with HAM/TSP, ATLL, IDH, and asymptomatic was 0.007, and the genetic distance values within and between the different clinical profiles are described in Table 2. The low overall genetic diversity found corroborates the fact that the HTLV-1 genome exhibits relatively few sequence variations and that the development of a therapeutic vaccine is possible. However, studies demonstrated that the induction of HTLV-1 protective immune response is not so simple. 12 –15 Here, we suggest that a therapeutic vaccine may be a better alternative and the HTLV-1 ORF-I is a good target for the development of this vaccine. More analyses involving sequences from patient with others HTLV-1 pathologies can provide more information about the ORF-I genetic diversity and these data can be used for a design of HTLV-1 vaccine.
Genetic Distances in Human T Cell Lymphotropic Virus Type 1 Open Reading Frame-I Sequences from Patients with Different Clinical Profiles
Footnotes
Acknowledgments
The authors are grateful to all participating donors, the professionals of the centers of care and the sequencing platform of FIOCRUZ / IGM. This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 400900/2013-0, CNPq 150892/2018-7).
Availability of Supporting Data
All sequences are available in the GenBank database (accession numbers MF158987-MF159019).
Author Disclosure Statement
No competing financial interests exist.