
Abstract
Human immunodeficiency virus-1 (HIV-1) causes CD4 T cell depletion through a number of mechanisms, including programmed cell death pathways (both apoptotic and nonapoptotic). In the setting of HIV-1 infection, the enhanced lymphocyte cell death occurs as a consequence of complex interactions between the host immune system and viral factors, which are reviewed herein. On the other hand, the main challenge to HIV-1 eradication is the development of latent infection in a subset of long lived cells, including CD4+ T cells and macrophages, which resist HIV-induced cell death. Understanding the potential mechanisms of how HIV-1 induces lymphocyte cell death is critical to the “kick and kill” cure strategy, which relies on the effective killing of reactivated, HIV-1-infected cells.
Introduction
Human immunodeficiency virus-1 (HIV-1) is a retrovirus that primarily infects CD4+ T lymphocytes from peripheral blood, lymphoid tissue, and gut-associated lymphoid tissue. More than 80% of HIV-1-infected adults acquire the virus through mucosal exposure, and the rest are infected by percutaneous or intravenous inoculation. 1 Primary mucosal HIV-1 transmission likely occurs through the Langerhans cell, a type of dendritic cell present in oral and genital mucosal surfaces, or by direct viral infection of CCR5+ CD4+ T cells located near mucosal surfaces. 2,3
The initial step of HIV-1 infection occurs when viral envelope glycoproteins [glycoprotein 120 (gp120) and gp41] interact with the CD4 receptor and CCR5 or CXCR4 co-receptors. A conformational shift leads to viral-cell membrane fusion, and viral contents (two RNA strands and three enzymes: integrase, protease, and reverse transcriptase) are released into the host cell cytoplasm. Reverse transcriptase mediates the transcription of viral RNA into double-stranded DNA, which then integrates into the host genome. Once integrated, the virus may remain quiescent for prolonged periods, or may reactivate stochastically, or given an appropriate stimulus. Once a reactivation stimulus is present, host transcription factors [e.g., nuclear factor kappa B (NF-κB), nuclear factors of activated T cells, and specificity protein 1] induce proviral DNA transcription, mediated by host RNA polymerase II and modified by viral activators [e.g., HIV-1 transactivator of transcription (Tat) accessory protein]. This generates viral proteins that assemble and are released as a viral particle from the host cell membrane, which then further matures and infects other cells.
After the primary or acute phase of HIV-1 infection (2–4 weeks), a state of clinical latency occurs. Most HIV-1-positive persons left untreated experience a gradual, but progressive CD4 T lymphocyte depletion over the course of 5–10 years. In the absence of antiretroviral treatment (ART), CD4 T cell counts eventually fall below 200 cells/mm3 with increased susceptibility to opportunistic infections and malignancies.
The depletion of CD4+ T lymphocytes in the absence of ART is mediated by a number of different pathways. The HIV-1 cytopathic effect was once considered to be the primary mechanism of CD4 T cell destruction. 4 Subsequent studies showed that CD4 T cell depletion and AIDS development involve more than direct viral killing. It is now understood that an arguably more important mechanism involves regulated pathways (apoptotic, but also nonapoptotic) resulting in morphological changes and death of infected and uninfected cells. 5 –7
Apoptosis, also known as programmed cell death or type I cell death, is characterized by cytoplasmic shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blebbing leading to the formation of small vesicles known as apoptotic bodies, which are engulfed by neighboring phagocytic cells and degraded by lysosomes. 8 Although apoptosis is a normal physiologic process that regulates cell turnover during development and health, altered regulation of apoptosis can have pathophysiologic consequences: too much causes tissue degeneration and too little is associated with cancer. In the setting of HIV-1 infection, the enhanced apoptosis of CD4 T cells occurs as a consequence of complex interactions between the host immune system and viral factors, and can occur through multiple overlapping and nonexclusive pathways.
The objective of this review article is to briefly describe these molecular pathways involved in apoptosis as well as delineate HIV-1-specific factors modulating apoptotic cell death. In addition, we describe some of the nonapoptotic regulated cell death pathways, which play a role in cell depletion during HIV-1 infection. We also highlight how modulating apoptotic death of latently infected cells could contribute to an HIV cure.
Regulation of Apoptosis
Classically, apoptosis can be initiated by external forces, for example, death receptor signaling, which causes extrinsic apoptosis, or by alteration of the intracellular milieu, which can cause apoptosis from within, or intrinsic apoptosis.
Extrinsic Pathway of Apoptosis
The extrinsic pathway is initiated through activation of death receptors after binding of specific ligands (Fig. 1). Following receptor ligation and trimerization, the intracellular death domains of the receptors recruit adapter proteins [e.g., Fas associated through death domain (FADD) and/or TNFRSF1A associated through death domain (TRADD)], which then recruit and activate caspase-8 or caspase-10 to initiate an apoptosis signaling cascade.
8,9
The ligands are unable to penetrate hydrophobic cell membranes; therefore, their signal must be transduced into cell by other mechanisms that comprise receptor activation.
10
These factors are grouped within the human tumor necrosis factor (TNF) superfamily: Extrinsic pathway of apoptosis. TNF superfamily comprises 19 ligand members that interact with 29 different receptors, which play a key role in different cell functions (proliferation, morphogenesis, and apoptosis). The factors specifically related to apoptosis are as follows (ligand/receptor): TNFα/TNFR1 and R2; TNFβ/TNFR1 (DR1); FasL/Fas; CD40L/CD40; CD30L/CD30; TNF-related apoptosis-inducing ligand (TRAIL)/TRAILR1, R2, R3, and R4; 4-1BBL/4-1BB; TNF weaker inducer of apoptosis (TWEAK)/TWEAKR; TNF superfamily member 14 (TNFSF14)/TNFSFR14; vascular endothelial cell growth inhibitor (VEGI)/DR3 and DcR3; ectodysplasin A1 (EDA-A1)/EDAR, and ectodysplasin A2 EDA-A2/XEDAR. The ligands are primarily transmembrane factors on the cell surface and interact with the cells that contain the respective receptors; however, some of them can be expressed as a soluble protein (TNFβ) and both as transmembrane and soluble form (TNFα).
11
Steps after TNF superfamily ligand/death receptor interaction: FasL/Fas interaction activates FADD, which then binds to the receptor death domain of and sequentially activates caspase-8. The multiprotein complex formed by the receptor death domain, FADD and caspase-8, is called death-inducing signaling complex (DISC).
10
Conformational changes of activated caspase-8 result in activation of the effector downstream caspases—caspase-3 and caspase-7—which in turn lead to cleavage of vital cellular proteins, such as structural proteins (lamins and gelsolin), as well as activation of DNase and consequent cell death. Caspase-8 activation can also cleave BH3 interacting domain death agonist (BID) into truncated BID (tBID), thereby activating a final common pathway with intrinsic apoptosis (see section “Intrinsic Pathway of Apoptosis”). It is important to note the key role of c-FLIP [cellular FLICE (FADD-like interleukin [IL]-1 beta-converting enzyme)-inhibitory protein] in this setting. The c-FLIP is a molecule with sequence homology to caspase-8 and has the ability to modulate caspase-8 activation. Once activated, c-FLIP also binds to the DISC complex.
8,9,12
TNF/TNFR1 interaction recruits TRADD, which interacts with receptor-interacting serine/threonine-protein kinase 1 (RIPK1). Depending on RIPK1 ubiquitylation or deubiquitylation, the TNF pathway outcome can be cell survival or apoptosis. Apoptosis requires deubiquitylation of RIPK1, which exists as a free molecule and assembles in the cytosol with TRADD, FADD, procaspase-8, and a long isoform of c-FLIP, generating autocatalytic cleavage and activation, releasing active caspase-8, and culminating in cell death.
13,14
TRADD can also initiate mitochondrial-dependent apoptosis through tBID.
Intrinsic Pathway of Apoptosis
The mitochondrion plays an indispensable role in intrinsic apoptosis. It is a double membrane-bound organelle found in the cytoplasm of eukaryotic cells. One of its main components is the outer mitochondrial membrane (OMM), which encloses the entire organelle and contains voltage-dependent anion channels, allowing the traffic of small molecules (e.g., ions and sugars). The inner mitochondrial membrane (IMM) folds toward itself and accommodates internally to the OMM. In normal conditions, the space between the IMM and OMM—called intermembrane space—has the same molecular concentration of the cytosol. Concentration of large proteins, although, is different as they need the help of “transporters' to navigate across the outer membrane; one example of this type of protein is cytochrome c. Transit of cytochrome c through the OMM is conducted by another group of proteins—belonging to the B cell lymphoma 2 (BCL2) family (see section “Category 2: BCL2 proapoptotic proteins”)—which are able to form pores in the OMM and increase its permeability. Once permeabilization takes place, cytochrome c is released to the cytosol and a cascade of molecular events culminate on caspase activation and apoptotic cell death 15 (Fig. 2).
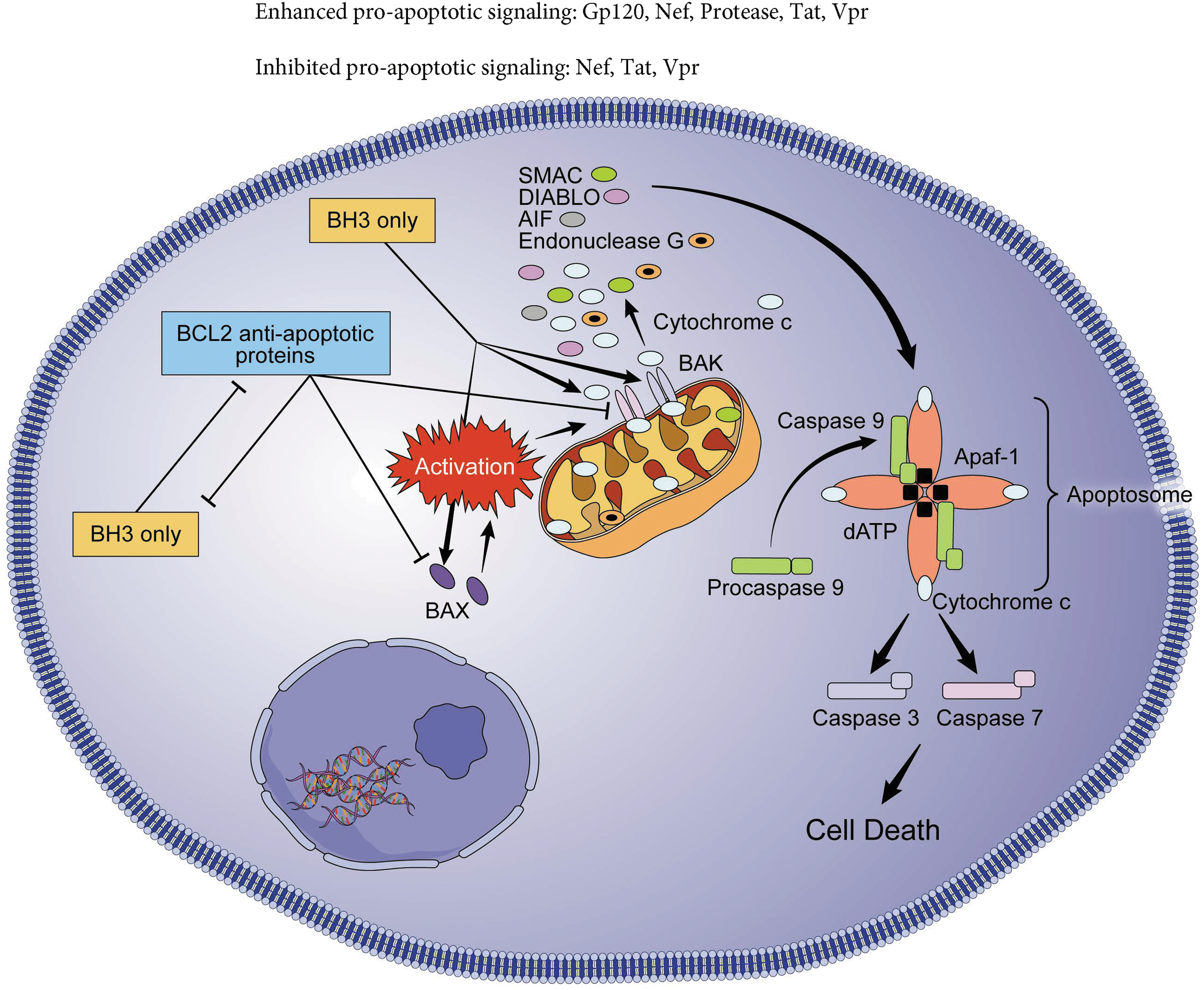
Intrinsic pathway of apoptosis. The permeabilization of the OMM induces the efflux of intermembrane proteins, which act as apoptogenic factors through pores formed by BAX and BAK. The first to be released is cytochrome c, although endonuclease G, AIF, SMAC/DIABLO, and HtrA2/OMI are also released. Cytochrome c binds to Apaf-1 and dATP, leading to activation of caspase-9, which also binds to the other factors and forms the apoptosome. The apoptosome activates caspase-3 and caspase-7 leading to DNA fragmentation and apoptosis. BCL2 antiapoptotic proteins execute antiapoptotic function by directly binding BAX and BAK or by sequestering their activators (BH3-only proteins). BH3-only proteins execute their proapoptotic function neutralizing the antiapoptotic BCL-2 proteins and also by direct activation of BAX and BAK. Effects of individual HIV proteins are annotated. OMM, outer mitochondrial membrane; BCL2, B cell lymphoma 2; BAK, BCL2 antagonist/killer 1; BAX, BCL2-associated X, apoptosis regulator; BOK, BCL2 family apoptosis regulator BOK; AIF, apoptosis-inducing factor; DIABLO, diablo IAP-binding mitochondrial protein; HTRA2, HtrA serine peptidase 2.
The intrinsic pathway of apoptosis is characterized by formation and opening of mitochondrial permeability transition pore in the OMM as well as loss of transmembrane potential and outer membrane permeabilization. 16 A variety of cytotoxic stimuli can activate the intrinsic pathway and this phenomenon is orchestrated by the interplay of proteins from BCL2 apoptosis regulator family, which contain both proapoptotic and antiapoptotic family members. These proteins share one to four BCL2 homology (BH) domains (BH1, BH2, BH3, and BH4). 8,17 They are categorized in the three groups below:
Category 1: BCL2 antiapoptotic proteins (the prosurvival proteins or “guardians”)
These proteins contain all four BH domains, are generally inserted into the OMM, and execute antiapoptotic functions by directly binding proapoptotic members of the BCL2 family (see section “Category 2: BCL2 proapoptotic proteins”) or by sequestering their activators (see section “Category 3: BCL2 apoptotic activator proteins”). The members of this family are as follows: BCL2 itself; BCL2-like 1 (BCL2L1 or BCL-XL); myeloid cell leukemia sequence 1 (MCL1); BCL2-like 2 (BCL2L2 or BCL-W); BCL2-related protein A1 (BCL2A1 or BFL-1); and BCL2-like 10 (BCL2L10 or BCL-B).
Category 2: BCL2 proapoptotic proteins
These members also harbor all four conserved BH domains. Three distinct proteins form this category: BCL2 antagonist/killer 1 (BAK); BCL2-associated X, apoptosis regulator (BAX); and BCL2 family apoptosis regulator BOK (BOK). BAX and BAK, once activated, insert themselves into the OMM as homo-oligomers, resulting in pore formation and promoting the release of apoptogenic proteins present in the intermembrane space of mitochondria. 18
BAX exists as inactive monomers on the cytosol. Once activated by apoptotic activators, conformational changes to BAX promote targeting, homo-oligomerization, and insertion into the mitochondrial OMM, which is essential for pore formation. 18
BAK is not only present at the OMM but also in the endoplasmic reticulum (ER). In contrast to BAX, BAK is constitutively inserted in the mitochondrial outer membrane (MOM); however, it also needs to be activated by some of the same BAX activators to promote pore formation. 19,20
BOK: BOK has been shown to induce mitochondrial outer membrane permeabilization (MOMP) and apoptosis in the absence of BAX and BAK and independent of other BCL2 family members. BOK proapoptotic activity is regulated by proteasome inhibition (which leads to BOK stabilization and apoptosis) and ER-associated protein degradation dysfunction. 17
Category 3: BCL2 apoptotic activator proteins
These proteins are also called BH3-only proteins since they share homology sequence only with the BH3 domain. 21 They carry out their proapoptotic function by neutralization of the prosurvival BCL2 family proteins or by direct activation of the proapoptotic effectors BAX and BAK. 17 BCL2-like 11 (BCL2L11 or BIM), BCL2 binding component 3 (BBC2 or PUMA), and tBID bind and inactivate all BLC2 antiapoptotic proteins [see “Category 1: BCL2 antiapoptotic proteins (the prosurvival proteins or “guardians” section)]. BCL2-associated agonist of cell death (BAD) only inactivates BCL2 itself, BCL-XL, and BCL-W, whereas phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1 or NOXA) just can neutralize MCL1 and BCL2A1.
Steps After Pore Formation and OMM Permeabilization
The permeabilization of the OMM induces the efflux of intermembrane proteins, which act as apoptogenic factors, including cytochrome c, endonuclease G, apoptosis-inducing factor mitochondria-associated 1 (AIFM1), diablo IAP-binding mitochondrial protein (DIABLO), and HtrA serine peptidase 2 (HTRA2). Cytochrome c binds to apoptotic peptidase activating factor 1 and dATP to form the apoptosome. The apoptosome activates caspase-9, leading to activation of caspase-3 and caspase-7 and the activation of DNA fragmentation factor subunit beta (DFFB or CAD), which translocates to the nucleus to induce DNA fragmentation. In addition, caspase-3 promotes chromatin condensation and this is contributed by endonuclease G and AIFM1. 22
Perforin/Granzyme Pathway
The perforin-/granzyme-dependent apoptotic cell death is an additional pathway by which cytotoxic T lymphocytes (CTL) and natural killer (NK) cells kill virally infected cells (Fig. 3). Granzymes are serine proteases released by cytoplasmic granules within the CTLs and NK cells. 23 There are two types of granzyme and each one differs slightly on how they induce apoptosis. Granzyme B, once released, enters the target cell and—together with another protein (perforin)—leads to activation of caspase-8, caspase-3, caspase-6, caspase-7, and caspase-9. However, Pinkoski et al. have demonstrated that granzyme B can induce also apoptosis in a caspase-independent route by cleaving and activating BID, inducing OMM permeabilization and cytochrome c release. 24 Granzyme A induces apoptosis in a completely caspase-independent manner by targeting the ER-associated Suppressor of Variegation, Enhancer of Zeste, and Trithorax (SET) complex. SET complex is generated together with superoxide when the mitochondria are damaged as a secondary event of granzyme A delivery to the cell. It is normally associated with the ER and is translocated to the nucleus of target cells interfering with DNA repair function. 25,26
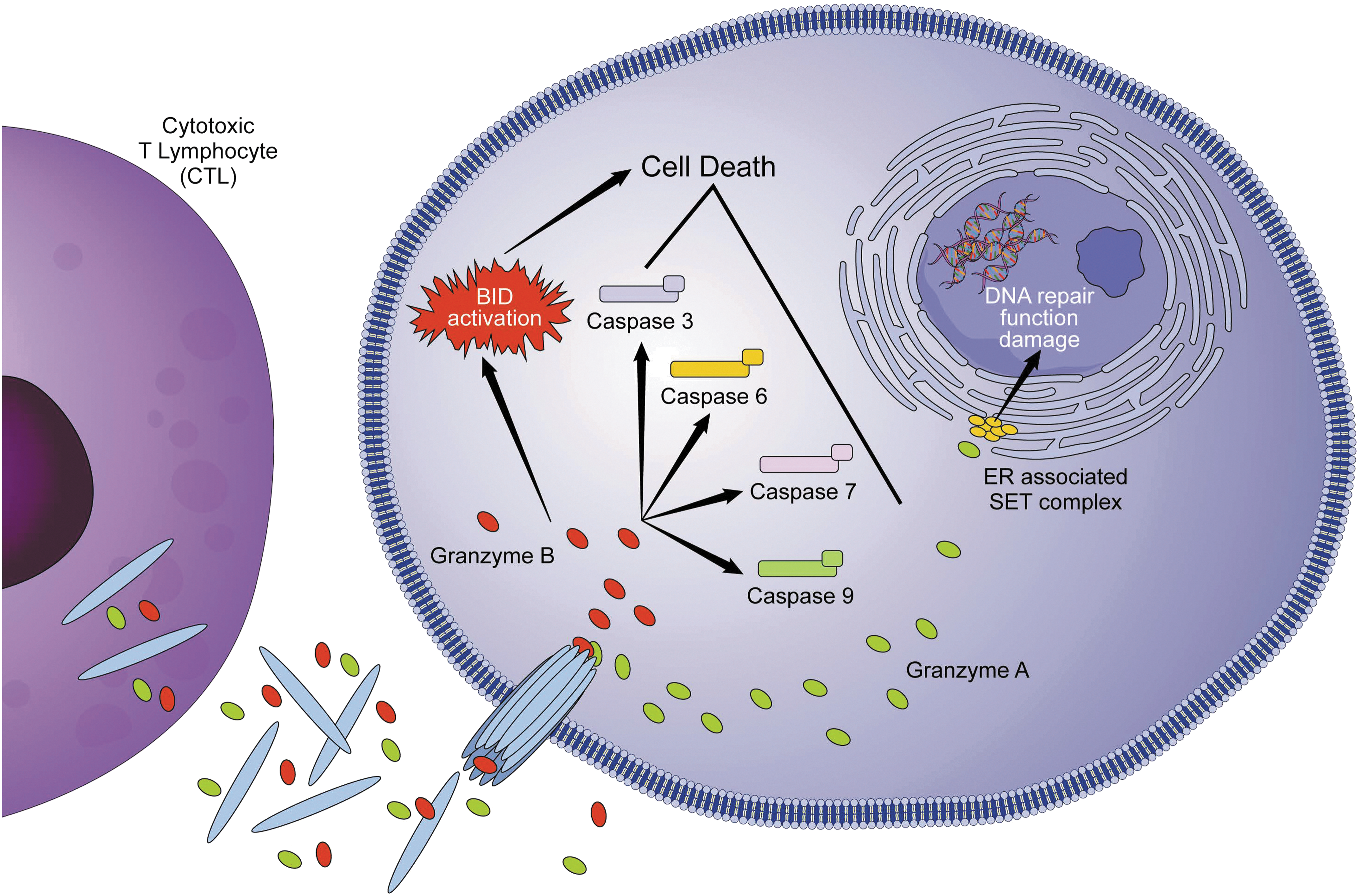
Perforin/granzyme pathway of apoptosis. Granzyme A and B are released from cytoplasmic granules within CTLs and NK cells. Granzyme B, once released, enters the target cell and leads to activation of caspase-8 as well as caspase-3, caspase-6, caspase-7, and caspase-9. It also can induce apoptosis by cleaving BID, inducing OMM permeabilization and cytochrome c release. Granzyme A targets the endoplasmic reticulum-associated SET complex, which translocates to the nucleus of target cells and interferes with DNA damage repair. CTL, cytotoxic T lymphocyte; NK, natural killer; SET, Suppressor of Variegation, Enhancer of Zeste, and Trithorax.
HIV Effects on Apoptosis
HIV-1 encodes several viral genes—three are common with all retroviruses (gag, pol, and env); two are regulatory genes (tat and rev); and four are accessory genes (vif, vpr, vpu, and nef). 27 Each of the viral proteins encoded by these genes has been shown to potentially modulate apoptosis (Table 1). In this section, the most studied HIV-1 proteins capable of modulating apoptotic cell death will be briefly reviewed. A comprehensive review of each individual HIV-1 protein, although, is beyond the scope of this artcle.
Proapoptotic and Antiapoptotic Effects of Individual Human Immunodeficiency Virus-1 Proteins
BCL2, B cell lymphoma 2; BAK, BCL2 antagonist/killer 1; BAX, BCL2-associated X, apoptosis regulator; TNF, tumor necrosis factor; Nef, negative regulatory factor; Vpr, viral protein R; c-FLIP, cellular FLICE (FADD-like interleukin-1 beta-converting enzyme)-inhibitory protein; SIRT1, sirtuin 1; TRAIL, TNF-related apoptosis-inducing ligand; gp120, glycoprotein 120; Tat, transactivator of transcription; Vpu, viral protein unique.
It should be noted at the outset, although, that many studies in this field rely on the use of exogenous overexpression of individual viral proteins, defective viral constructs deficient in one or more viral genes, and/or immortalized cell lines, which may not be reflective of true biologic phenomenon. In fact, depending on the virus life cycle, host cell type, and activation state, individual viral proteins can act as mediators of proapoptotic or antiapoptotic signals. Therefore, careful attention to these important details is necessary when evaluating reports on direct viral effects on cell death pathways.
Glycoprotein 120
HIV-1 gp120, encoded by env, is an envelope glycoprotein that resides in the outer membrane of the virus, can be shed from mature virions into the circulation and lymphoid tissue, and is expressed on the surface of infected cells. 28 It binds to the CD4 receptor and chemokine co-receptors (CXCR4 and CCR5) facilitating viral attachment. The serum forms of gp120 activate B cells resulting in production of anti-gp120 antibodies and formation of immune complexes. 29,30 The effects of gp120 on apoptosis are pleiotropic. For instance, exposure of CD4 T cells to gp120 increases susceptibility to apoptosis mediated by Fas 31 and TRAIL, 32,33 and stimulates TNFα production. 34 In addition, it can lead to reduced expression of BCL2 in CD4 T cells, 35 and increased expression of BCL2 binding component 3 (BBC3 or PUMA), one of the BCL2 apoptotic activator proteins. 36 Envelope glycoproteins gp120 and gp41 expressed on the surface of infected cells can interact with CD4 and/or chemokine co-receptors on nearby uninfected cells, inducing apoptosis and also cell-to-cell fusion with the formation of multinucleated syncytia and apoptosis by intrinsic apoptotic pathway. 37 –39
Proapoptotic effects of gp120 are not limited to CD4 T cells. For instance, either virion-bound gp120 or circulating immune complexes can sensitize monocyte-derived dendritic cells to undergo apoptosis in response to CD40L/CD40 ligation, which typically occurs after the cell migration into the lymphoid tissue, through increased expression of mitogen-activated protein kinase kinase kinase 5 (MAP3K5 or ASK-1), 40 a key intermediate in the Fas and TNFα death signaling cascades that leads to translocation of BAX to the mitochondria from the cytosol. 41
Negative regulatory factor
Negative regulatory factor (Nef) is an accessory HIV-1 protein, which is also present in simian immune deficiency virus. 42 It activates NF-κB, with consequent increase in the expression of HIV-1 genome as well as inflammatory factors, including IL-6. These factors will lead to activation of signal transducer and activator of transcription 3, which induces gene expression, cell survival, cell activation, and high-level viral replication. 43,44 Teleologically, then, it is no wonder that Nef has been described to have antiapoptotic effects in CD4 T cells. Nef [and viral protein unique (Vpu)]-mediated downregulation of surface CD4 expression on T cells may protect against antibody-mediated cellular cytotoxicity. 45 Expression of Nef in T cells induces phosphorylation and inactivation of proapoptotic BAD. 46 Furthermore, direct binding of Nef to ASK1 inhibits its kinase activity and proapoptotic signaling in T cells. 47,48 The physiologic relevance of these findings is supported by a recent study, which showed that inhibition of the Nef-ASK1 interaction increased CD8 T cell killing of autologous CD4 T cells from HIV-infected patients. 49
Conversely, expression of Nef may also promote apoptosis. Endogenous overexpression of HIV Nef in cell lines can sensitize to intrinsic apoptosis through downmodulation of BCL2 and BCLXL. 50 However, transfection of activated CD4 T cells with Nef induces lysosomal permeabilization with release of cathepsin-D, OMM rupture, and cell death. 51 Nef can be secreted by HIV-infected cells in exosomes and induces apoptosis in bystander cells, 52 the significance of which has been evaluated in a mouse model of HIV. 53
Protease
Protease is one of the three HIV-1 pol gene proteins (the other two being integrase and reverse transcriptase) the main function of which is to process gag-pol proteins to promote viral maturation. 54 Strack et al. first demonstrated in 1996 that HIV-1 protease also cleaves BCL2, in vitro and in cell-free extracts, leading to apoptosis. 55 Importantly, in that study, HIV-1 protease-mediated apoptosis was abrogated by endogenous overexpression of BCL2. Another more recently proposed mechanism by which protease induces apoptosis is the cleavage of procaspase-8, generating a novel Casp8p41 fragment, which directly binds and activates BAK causing mitochondrial-dependent apoptosis. 56 –60 This protease generated Casp8p41 cleavage fragment has been demonstrated in in vitro-infected cells, ex vivo patient cells, and patient lymph node biopsies, suggesting biologic relevance, 61,62 and is inhibited by BCL2. 63 Interestingly, Casp8p41 expression in T cells also enhances HIV replication by activating NF-κB-dependent HIV-1 long terminal repeat (LTR) activation. 64
Transactivator of transcription
HIV-1 Tat is produced early after viral gene transcription, and contributes significantly to viral replication and HIV-1 pathogenesis. 65 The way Tat modulates apoptosis may depend on Tat levels and whether the cell is HIV infected or not. On the one hand, Tat expression in primary CD4 T cells decreases cell susceptibility to Fas and increases the expression of BCL2 and c-FLIP in transfected Jurkat T cells. 66 HIV-produced Tat can also neutralize lysine acetyltransferase 5 (KAT5 or Tip60), a type of histone acetyltransferase, reducing Tip60-dependent apoptotic cell response to DNA damage. 67
On the other hand, multiple proapoptotic effects have been attributed to Tat. HIV-produced Tat has been shown to increase procaspase-8 expression in Jurkat T cells, increasing susceptibility to Fas, 68 and to increase susceptibility to HIV gp120-induced apoptosis. 31 Tat also promotes cell microtubule alteration, inducing apoptosis through mitotic arrest. 69 –71 Tat stimulates transcription of cyclin B1, an important cell cycle regulator, which controls mitosis and when overexpressed can sensitize cells to apoptosis induction. 72 In addition, Tat can also promote cell death by inhibition of sirtuin 1, an NAD-dependent deacetylase, which removes acetyl groups from various proteins, and this interaction activates the tumor suppressor protein p53, which leads to T cell depletion. 73
Viral protein R
Viral protein R (Vpr) is a multifunctional protein essential for efficient viral infection. Vpr is involved in nuclear transport of the HIV-1 preintegration complex and induction of G2 cell cycle arrest, which increases viral replication (since transcriptional activity of HIV-1 LTR is most active in G2 phase) and promotes apoptosis. 74 –76 Sato et al. 77 demonstrated that this effect occurs predominately in regulatory CD4+ T cells (Tregs) in vivo using the humanized mouse model. However, macrophages seem to be resistant to Vpr-induced apoptosis due to expression of baculoviral IAP repeat-containing proteins. 78 A potential mechanism for Vpr-induced apoptosis may be through a direct interaction of Vpr with the permeability transition pore complex, a mitochondrial pore bridging the IMM and OMM, leading to membrane permeabilization and release of proapoptotic proteins, 79 an effect that is inhibited by BCL2. In fact, a specific mutation of Vpr (R77Q mutation), which is overrepresented in HIV long-term nonprogressors, results in less apoptosis in vitro compared to wild-type Vpr, 80 suggesting that Vpr-induced apoptosis has pathophysiologic consequences. More recently, it has been shown that Vpr expression in infected cells increase TNFα release, which, in turn, may impact apoptosis regulation. 81
Viral protein unique
Vpu is an HIV-1-encoded protein, which has a primary biological function of facilitating the viral release from infected cells. 82 It antagonizes bone marrow stromal cell antigen 2 (BST2 or tetherin), which prevents budding of virus particles from infected cells. 83 Overexpression of Vpu in T cell lines increases susceptibility to Fas-mediated apoptosis, whereas cells infected with viral clones that are Vpu deficient are less susceptible to Fas. 84 This may occur through inhibition of NF-κB-dependent expression of antiapoptotic proteins, including BCL2 family proteins or TNF-R complex proteins (e.g., TNF receptor-associated factor 1). 85 More recently, Park et al. demonstrated that overexpression of Vpu can result in caspase-mediated cleavage of interferon regulatory factor 3, a transcription factor that plays an important role in antiviral immune response, coincident with apoptosis, suggesting an immune escape advantage for inducing apoptosis in an infected cell. 86
HIV-1 infection of CD4 T cells can result in apoptosis by mechanisms unrelated to expression of individual HIV proteins as well. Cooper et al. demonstrated a potential mechanism of apoptosis dependent upon HIV-1 integration. 87 Viral DNA integration into the host cell genome can elicit a cellular double-stranded DNA damage response by activating DNA-dependent protein kinase (DNA-PK). Depending on specific autophosphorylation of DNA-PK and its localization during the cell cycle, it can lead to repair of DNA damage or induction of apoptosis. 88 The biologic significance of this potential pathway has remained controversial, 89 –91 as it is evident that not all HIV-1-infected cells that undergo integration also undergo apoptosis, or else the viral life cycle would be self-limiting and the latent reservoir would essentially not exist.
HIV-1 Infection and Nonapoptotic Cell Death Pathways
Over the last decade, an increasing body of literature has revealed nonapoptotic mechanisms involved in regulated cell death in the context of HIV-1 infection. The progressive refinement of research techniques has been able to demonstrate viral-induced cell depletion even in the lack of apoptotic elements. This has led to the discovery of a whole new array of factors orchestrating novel modalities of regulated cell death. The nonapoptotic pathways include necroptosis, pyroptosis, and autophagy.
Necroptosis
Necroptosis is a form of regulated cell death, which is typically triggered by pathogen invasion and is mediated by the interaction between ligand with its death receptor in an analogous way to extrinsic apoptotic pathway. 8 Some apoptotic ligands (and their respective receptors) can induce necroptosis, for example, Fas, TNF, and TRAIL. However, Toll-like receptor 3 (TLR 3) and TLR 4 have a necroptotic role as well 92 –94 (Fig. 4).
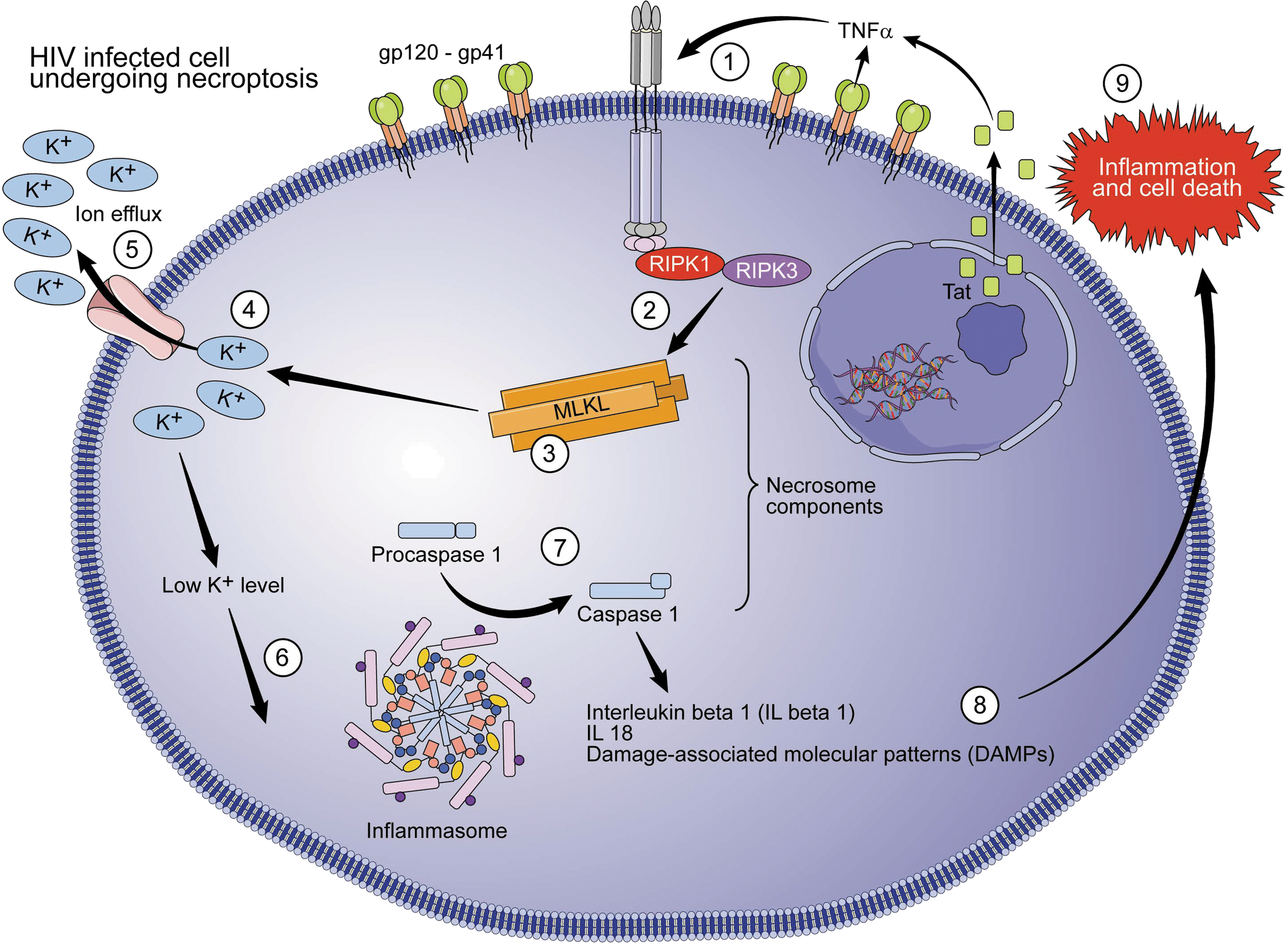
HIV-1 and necroptosis. Necroptosis pathway is started by interaction between TNF/TNFR1, leading to RIPK3 activation by RIPK1. Active RIPK-3 catalyzes the phosphorylation of MLKL, resulting in MLKL oligomers, which insert themselves in the plasma membrane, forming an ion channel and triggering plasma membrane permeabilization. The ionic dysregulation promotes the formation of the inflammasome, which leads to activation of caspase-1 and consequent secretion of mature IL-β1, IL-18, and DAMPs. The final products are inflammatory response and necrotic cell morphology with cell swelling, diffuse fragmentation, and membrane integrity loss. The HIV-1 Tat protein leads to increased TNFα production and necroptosis stimulation. MLKL, mixed-lineage kinase domain-like pseudokinase; IL, interleukin; DAMP, damage-associated molecular pattern; Tat, transactivator of transcription.
At the molecular level, necroptosis is mediated by the sequential activation of receptor-interacting serine/threonine kinase 3 (RIPK3) and mixed-lineage kinase domain-like pseudokinase (MLKL). Both factors together are called the necrosome. Once TNF/TNFR1 interaction takes place, RIPK3 is activated by RIPK1. Active RIPK3 catalyzes the phosphorylation of MLKL, resulting in MLKL oligomers, which insert themselves in the plasma membrane, forming an ion channel and triggering plasma permeabilization. The ionic dysregulation promotes the formation of the inflammasome, a supramolecular platform that leads to activation of caspase-1 and consequent secretion of mature IL-1β, IL-18, and damage-associated molecular patterns (DAMPs) or alarmins. The final product is an inflammatory response and necrotic cell morphology with cell swelling, diffuse fragmentation, and membrane integrity loss. 95,96
In the setting of HIV-1 infection, Pan et al. recently demonstrated that at least some HIV-induced cell death in vitro can be inhibited by necrostatin-1, a RIPK1 inhibitor, which blocks necroptosis. 97 The in vivo relevance of this observation is unclear and more work needs to be done in this area. However, inhibition of necroptosis and knockdown of RIPK3 restored proliferative capacity of ex vivo HIV-specific CD8 T cells from HIV-positive patients, suggesting this form of cell death may contribute to HIV-induced immunosuppression in ways other than through CD4 T cell depletion. 98
Pyroptosis
Pyroptosis is a form of regulated cell death, which relates to innate immunity and is mainly triggered by intracellular pathogen invasion (Fig. 5). In regard of cell morphology, it has some similarities with apoptosis (chromatin condensation) and necrosis (cellular swelling, culminating with membrane integrity loss). 8,99 Another key difference from apoptosis is the activation of caspase-1. Recognition and internalization of pathogen-associated molecular patterns (PAMPs) by TLRs expose the external antigens to the intracellular environment and to a second line of receptors, Nod-like receptors. This leads to activation of caspase-1, which further activates caspase-3, caspase-4, and caspase-5 and orchestrates the formation of a multiprotein intracellular complex called the inflammasome. Inflammasome activates proinflammatory cytokines IL-1β and IL-18, which travel to the extracellular environment once the intracellular protein, Gasdermin D, is cleaved in two subunits, which will be inserted in the cellular membrane and form pores. 100,101
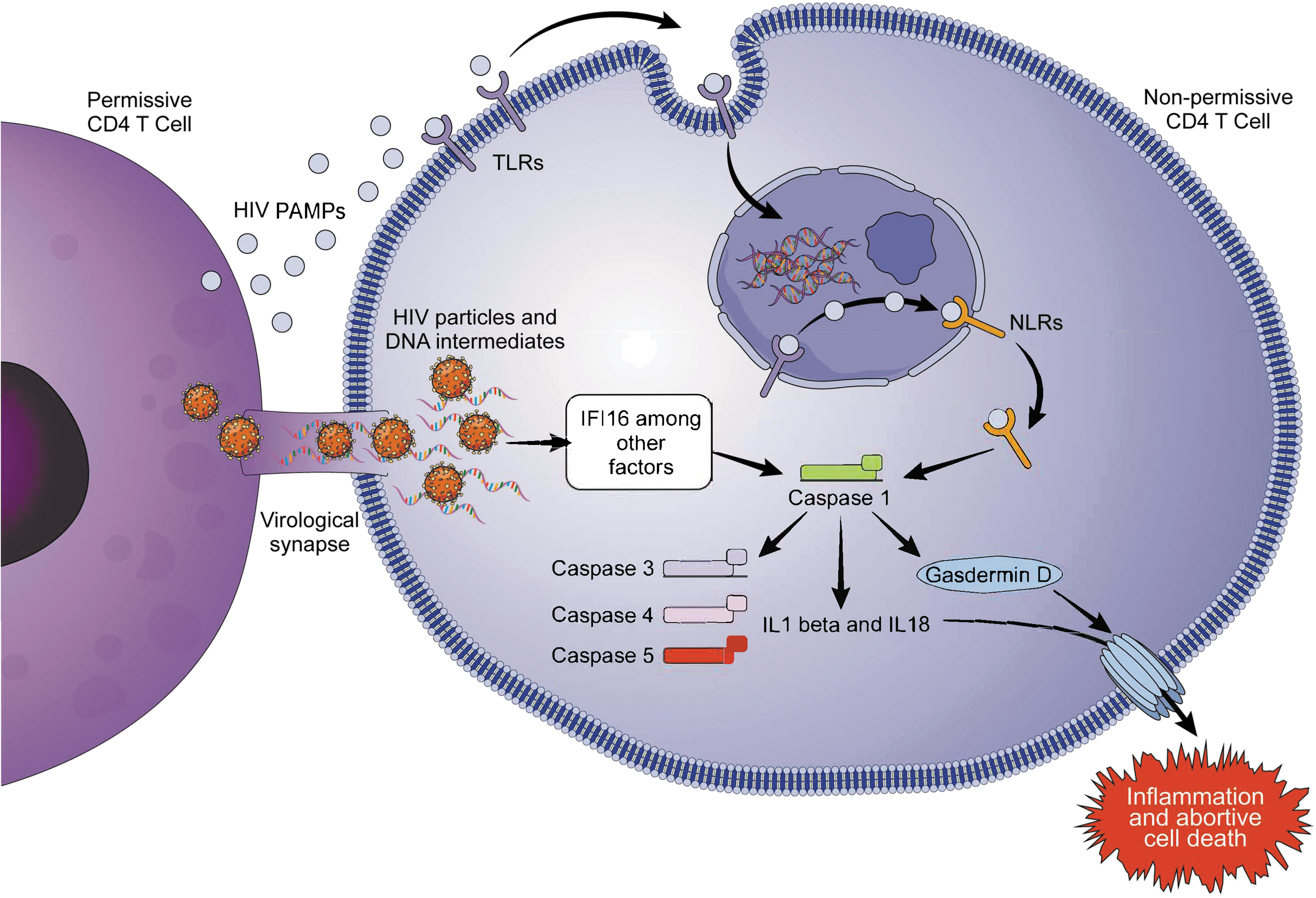
HIV-1 and pyroptosis. In quiescent lymphoid cells, pyroptosis is induced by the accumulation of viral DNA intermediates transferred from actively infected (permissive) CD4+ T cells through cell-to-cell spread (virological synapse). The initial step of the pyroptotic pathway is the recognition of the PAMPs by TLRs. They expose the external antigen to the intracellular environment and to a second line of receptors, NLRs. Next step is activation of caspase-1, which further activates caspase-3, caspase-4, and caspase-5 and increases secretion of proinflammatory cytokines IL-1β and IL-18, which travel to the extracellular environment once the intracellular protein, Gasdermin D, is cleaved in two subunits, which will be inserted in the cellular membrane and form pores. Final products are inflammation and cell death. PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor; NLR, Nod-like receptor.
Doitsh et al. first demonstrated that pyroptosis plays a role in CD4 T cell death in the setting of HIV-1 infection. 102 They infected fresh human tonsil/spleen tissues with a CXCR4 tropic stain of HIV-1 and determined the distribution of caspase-1 and caspase-3 using fluorescently labeled inhibitor of caspases as surrogates of pyroptosis and apoptosis, respectively. Caspase-3 activity was identified in the productively infected CD4 T cells, whereas caspase-1 was identified dying nonpermissive CD4 T cells (cells that do not generate productive infection once HIV-1 is internalized). 103 In quiescent lymphoid cells, pyroptosis is induced by the accumulation of viral DNA intermediates transferred from actively infected (permissive) CD4+ T cells through cell-to-cell spread (virological synapse). 102,104 The DNA intermediate sensing is mediated by interferon gamma-inducible protein 16, which is one of the mediators of caspase-1 activation and pyroptotic cell death. 105 In contrast, blood-derived CD4 T cells are constitutionally resistant to caspase-1-dependent regulated cell death. 106 HIV-induced pyroptosis does not seem to be a protective mechanism; in turn, it may create a vicious cycle when the release of inflammatory signals attracts more cells to die in the lymphoid tissue.
Autophagy
Autophagy is a degradation pathway characterized by the formation of double membrane-bound compartments, termed autophagosomes, which engulf and sequester cytoplasmic constituents (e.g., dysfunctional organelles, protein aggregates, and microbial pathogens) (Fig. 6). Degradation happens by fusion of autophagosomes with lysosomes. 107 Autophagy-dependent cell death is characterized by extensive cytoplasmic vacuolization leading to phagocytic uptake and lysosomal degradation. 8 This self-digestion pathway plays an important role in innate and adaptive immune response to pathogens, including viral infection. When microbial infection takes place, the autophagic pathway delivers microbial and viral products and derivatives (PAMPs and DAMPs) to the recognition of TLRs and major histocompatibility complex class II on macrophages and dendritic cells. Autophagy has two spectrums: (1) it can be an adaptive response to survival avoiding disruption of intracellular environment or (2) it can lead to cell death.

HIV-1 and autophagy. Autophagic pathway starts with the formation of double membrane-bound compartments, termed autophagosomes, which engulf and sequester cytoplasmic constituents (e.g., dysfunctional organelles, protein aggregates, and microbial pathogens). Degradation happens by fusion of autophagosomes with lysosomes. The HIV-1-encoded Nef protein inhibits autophagy by binding and sequestering beclin-1. It also inhibits lysosomal activity through phosphorylation of TFEB protein. Envelope glycoproteins (gp120 and gp41) activate beclin-1 (and autophagy) in bystander cells, but inhibits beclin-1 in HIV-1 infected CD4 T cells. Vpr protein is also able to induce autophagy and beclin-1 expression. HIV-1 Tat protein is degraded by autophagy, generating decreased viral transcription and replication. TFEB, transcriptor factor EB; Nef, Negative regulatory factor; Vpr, viral protein R; gp120, glycoprotein 120.
HIV-1-infected CD4+ T cells expressing surface envelope glycoproteins (gp120 and especially gp41 with its fusogenic activity) can induce autophagy in bystander CD4+ T lymphocytes. 108 This effect on bystander cells may be dependent upon the fusogenic activity of gp41, 109 and independent of co-receptor tropism. 110
However, productively infected CD4 T cells and macrophages, although, seem to be resistant to HIV-induced autophagy-dependent cell death. 110,111 Transcription factor EB (TFEB) is the main regulator of the expression of lysosomal hydrolases, membrane proteins, and genes involved in autophagic cell clearance. 112,113 The HIV-1-encoded protein Nef inhibits autophagy in macrophages by binding beclin-1, resulting in TFEB phosphorylation and cytosolic sequestration. 114,115 This protects HIV from degradation and contributes to a more efficient HIV-1 replication.
Conversely, non-cell death-inducing autophagy may be protective to the host. For instance, HIV-1 Tat has been shown to bind to sequestosome 1 (SQSTM1), leading to degradation by autophagy. 116 Similarly, immortalized monocyte cells transfected with HIV-1 Vpr degrade that protein through autophagy. 117 This would result in HIV restriction and decreased viral replication. A complete discussion on the role of non-cell death-inducing autophagy in HIV-1 infection is beyond the scope of this article, but has been recently reviewed elsewhere. 118
HIV-1-Induced Apoptosis and Implications for Possible HIV Cure
HIV-1 infection of CD4+ T cells (as well as some other hematopoietic cells, including macrophages) results in an array of events that are directly linked with chronic immunologic activation, active viral replication, and lymphocyte depletion. Several viral-encoded proteins (envelope proteins, protease, Nef, Tat, Vpr, and Vpu) have been robustly demonstrated to modulate the immune response, either favoring viral replication or promoting apoptotic cell death.
One of the main challenges to HIV-1 eradication is the development of latent infection in a subset of CD4+ T cells (resting central memory T cells and transitional memory T cells) and other cell types such as macrophages and naive T cells. 119 –121 These cells contain the viral DNA integrated to their genome, but they are not productively infected so they can silently harbor the virus for prolonged periods of time. Finzi et al. demonstrated that latently infected cells (or reservoirs) are very stable and have a half-life of more than 43 months. 122 These reservoirs also may have less sensitivity to apoptotic cell death mechanisms, for example Fas/FasL system, resulting in survival even in the setting of adequate HIV-1 suppression after effective ART. 123,124
One important approach in HIV-1 cure research involves the use of latency-reversing agents to reactivate virus and cause selective cell death of the infected cells—the “kick and kill” hypothesis. 125 Further understanding of how HIV-infected cells can die, and why some infected cells do not die, can inform strategies to modulate apoptosis and other signaling pathways to promote death of reactivated cells. 126 Indeed, early success in increasing cell death in reactivated, infected cells in vitro and ex vivo has been shown by targeting the DExD/H-box helicase 58 (DDX58 or RIG-1) pathway 127 ; the intrinsic apoptosis pathway 63,128,129 ; the extrinsic apoptosis pathway 130 ; and autophagy-dependent cell death. 131 These strategies will likely generate better results when effective HIV-1-specific cytotoxic T cell response is activated concomitantly. 132
One of the several challenges remaining is to refine techniques to identify and modulate cell death sensitivity in latently infected cells, specifically with minimal effects on uninfected cells. Furthermore, an increasing number of studies have been demonstrating that nonapoptotic forms of regulated cell death can be directly related to HIV-1 infection. It is yet to be determined how closely these pathways (e.g., necroptosis, proptosis, and autophagy) relate to apoptosis and if they work as compensatory or alternative cell death mechanisms. It would be of similar importance to understand in more detail the nonapoptotic cell death factors that can be potentially targeted and how they can function as adjunctive immunotherapy strategies toward HIV-1 eradication.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Mayo Clinic Foundation, and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R56AI145407 (N.W.C.), and R01AI110173 and R01AI120698 (A.D.B.).